

Chk1/2 inhibition overcomes the cisplatin resistance of head and neck cancer cells secondary to the loss of functional p53
- PMID: 23839309
- PMCID: PMC3955083
- DOI: 10.1158/1535-7163.MCT-13-0157
Chk1/2 inhibition overcomes the cisplatin resistance of head and neck cancer cells secondary to the loss of functional p53
Abstract
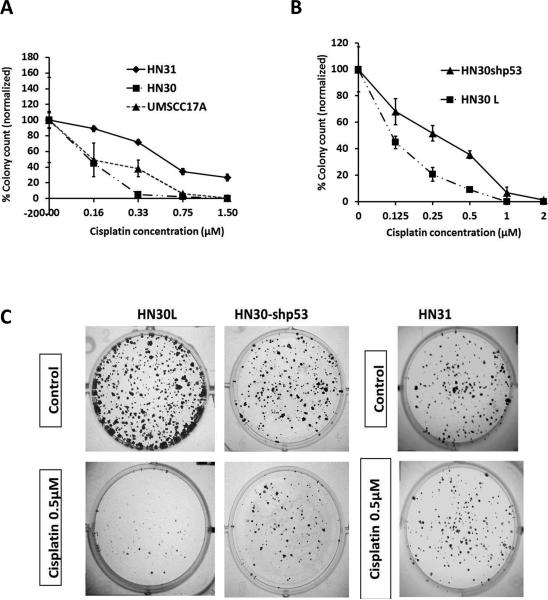
Despite the use of multimodality therapy using cisplatin to treat patients with advanced stage squamous cell carcinoma of the head and neck (HNSCC), there is an unacceptably high rate of treatment failure. TP53 is the most commonly mutated gene in HNSCC, and the impact of p53 mutation on response to cisplatin treatment is poorly understood. Here, we show unambiguously that wild-type TP53 (wtp53) is associated with sensitivity of HNSCC cells to cisplatin treatment, whereas mutation or loss of TP53 is associated with cisplatin resistance. We also show that senescence is the major cellular response to cisplatin in wtp53 HNSCC cells and that cisplatin resistance in p53-null or -mutant TP53 cells is due to their lack of senescence. Given the dependence on checkpoint kinase (Chk)1/2 kinases to mediate the DNA damage response in p53-deficient cells, there is potential to exploit this to therapeutic advantage through targeted inhibition of the Chk1/2 kinases. Treatment of p53-deficient HNSCC cells with the Chk inhibitor AZD7762 sensitizes them to cisplatin through induction of mitotic cell death. This is the first report showing the ability of a Chk kinase inhibitor to sensitize TP53-deficient HNSCC to cisplatin in a synthetic lethal manner, which has significance given the frequency of TP53 mutations in this disease and because cisplatin has become part of standard therapy for aggressive HNSCC tumors. These preclinical data provide evidence that a personalized approach to the treatment of HNSCC based on Chk inhibition in p53-mutant tumors may be feasible.
Figures





Similar articles
-
Wee-1 kinase inhibition overcomes cisplatin resistance associated with high-risk TP53 mutations in head and neck cancer through mitotic arrest followed by senescence.Mol Cancer Ther. 2015 Feb;14(2):608-19. doi: 10.1158/1535-7163.MCT-14-0735-T. Epub 2014 Dec 10. Mol Cancer Ther. 2015. PMID: 25504633 Free PMC article.
-
Mutations of the LIM protein AJUBA mediate sensitivity of head and neck squamous cell carcinoma to treatment with cell-cycle inhibitors.Cancer Lett. 2017 Apr 28;392:71-82. doi: 10.1016/j.canlet.2017.01.024. Epub 2017 Jan 23. Cancer Lett. 2017. PMID: 28126323 Free PMC article.
-
Targeting Chk1 in p53-deficient triple-negative breast cancer is therapeutically beneficial in human-in-mouse tumor models.J Clin Invest. 2012 Apr;122(4):1541-52. doi: 10.1172/JCI58765. Epub 2012 Mar 26. J Clin Invest. 2012. PMID: 22446188 Free PMC article.
-
TP53 Mutations in Head and Neck Squamous Cell Carcinoma and Their Impact on Disease Progression and Treatment Response.J Cell Biochem. 2016 Dec;117(12):2682-2692. doi: 10.1002/jcb.25592. Epub 2016 Jun 3. J Cell Biochem. 2016. PMID: 27166782 Free PMC article. Review.
-
[Molecular mechanisms of chemoresistance in head and neck squamous cell carcinoma].Lin Chuang Er Bi Yan Hou Tou Jing Wai Ke Za Zhi. 2017 Jun 5;31(11):888-891. doi: 10.13201/j.issn.1001-1781.2017.11.018. Lin Chuang Er Bi Yan Hou Tou Jing Wai Ke Za Zhi. 2017. PMID: 29775010 Review. Chinese.
Cited by
-
Phase I Study of LY2606368, a Checkpoint Kinase 1 Inhibitor, in Patients With Advanced Cancer.J Clin Oncol. 2016 May 20;34(15):1764-71. doi: 10.1200/JCO.2015.64.5788. Epub 2016 Apr 4. J Clin Oncol. 2016. PMID: 27044938 Free PMC article. Clinical Trial.
-
Cotargeting CHK1 and PI3K Synergistically Suppresses Tumor Growth of Oral Cavity Squamous Cell Carcinoma in Patient-Derived Xenografts.Cancers (Basel). 2020 Jun 29;12(7):1726. doi: 10.3390/cancers12071726. Cancers (Basel). 2020. PMID: 32610557 Free PMC article.
-
Cisplatin generates oxidative stress which is accompanied by rapid shifts in central carbon metabolism.Sci Rep. 2018 Mar 9;8(1):4306. doi: 10.1038/s41598-018-22640-y. Sci Rep. 2018. PMID: 29523854 Free PMC article.
-
Targeting the DNA Damage Response for Cancer Therapy.Int J Mol Sci. 2023 Nov 2;24(21):15907. doi: 10.3390/ijms242115907. Int J Mol Sci. 2023. PMID: 37958890 Free PMC article. Review.
-
Small Cell Lung Cancer: Emerging Targets and Strategies for Precision Therapy.Cancers (Basel). 2023 Aug 8;15(16):4016. doi: 10.3390/cancers15164016. Cancers (Basel). 2023. PMID: 37627044 Free PMC article. Review.
References
-
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
