

Genome-wide profiling identifies a DNA methylation signature that associates with TET2 mutations in diffuse large B-cell lymphoma
- PMID: 23831920
- PMCID: PMC3856967
- DOI: 10.3324/haematol.2013.088740
Genome-wide profiling identifies a DNA methylation signature that associates with TET2 mutations in diffuse large B-cell lymphoma
Abstract
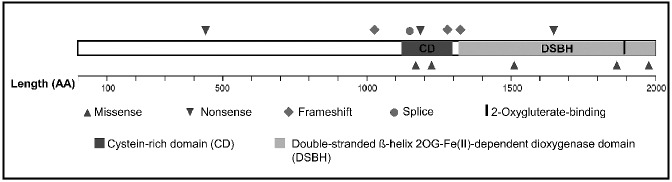
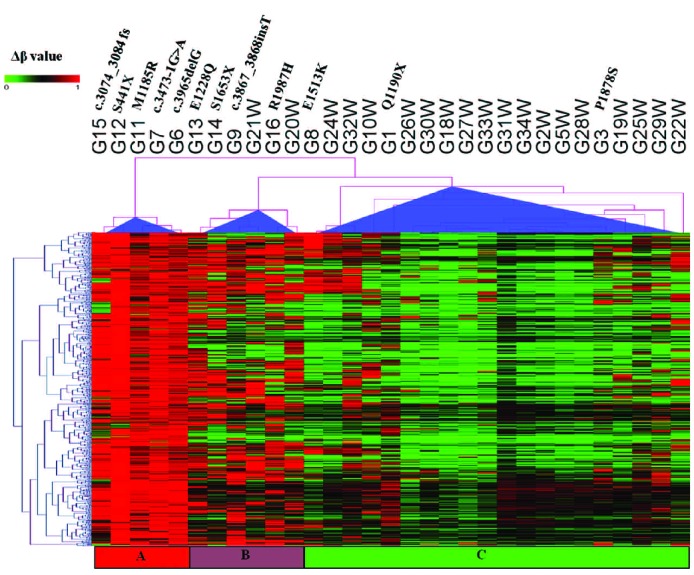
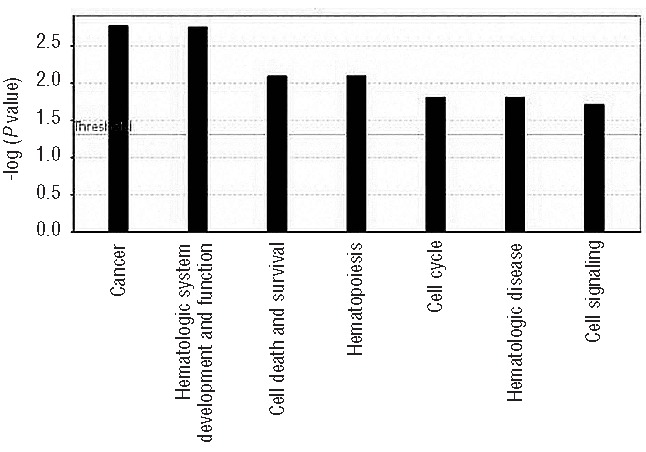
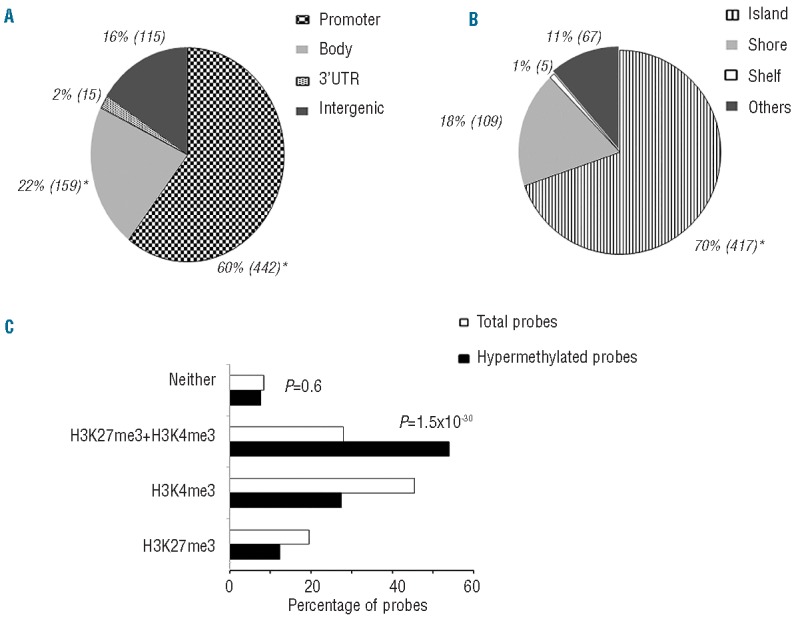
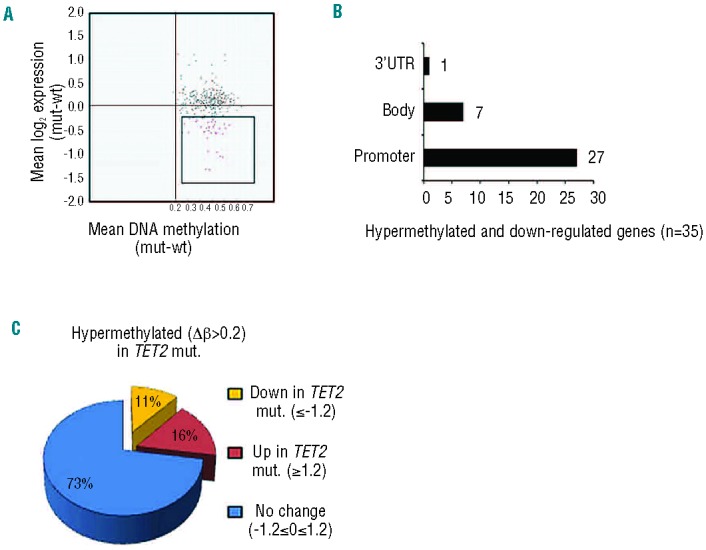
The discovery that the Ten-Eleven Translocation (TET) hydroxylases cause DNA demethylation has fundamentally changed the notion of how DNA methylation is regulated. Clonal analysis of the hematopoetic stem cell compartment suggests that TET2 mutations can be early events in hematologic cancers and recent investigations have shown TET2 mutations in diffuse large B-cell lymphoma. However, the detection rates and the types of TET2 mutations vary, and the relation to global methylation patterns has not been investigated. Here, we show TET2 mutations in 12 of 100 diffuse large B-cell lymphomas with 7% carrying loss-of-function and 5% carrying missense mutations. Genome-wide methylation profiling using 450K Illumina arrays identified 315 differentially methylated genes between TET2 mutated and TET2 wild-type cases. TET2 mutations are primarily associated with hypermethylation within CpG islands (70%; P<0.0001), and at CpG-rich promoters (60%; P<0.0001) of genes involved in hematopoietic differentiation and cellular development. Hypermethylated loci in TET2 mutated samples overlap with the bivalent (H3K27me3/H3K4me3) silencing mark in human embryonic stem cells (P=1.5×10(-30)). Surprisingly, gene expression profiling showed that only 11% of the hypermethylated genes were down-regulated, among which there were several genes previously suggested to be tumor suppressors. A meta-analysis suggested that the 35 hypermethylated and down-regulated genes are associated with the activated B-cell-like type of diffuse large B-cell lymphoma in other studies. In conclusion, our data suggest that TET2 mutations may cause aberrant methylation mainly of genes involved in hematopoietic development, which are silenced but poised for activation in human embryonic stem cells.
Figures
Similar articles
-
Integrated analysis of genome‑wide gene expression and DNA methylation microarray of diffuse large B‑cell lymphoma with TET mutations.Mol Med Rep. 2017 Oct;16(4):3777-3782. doi: 10.3892/mmr.2017.7058. Epub 2017 Jul 21. Mol Med Rep. 2017. PMID: 28731140 Free PMC article.
-
TET2 Mutations Affect Non-CpG Island DNA Methylation at Enhancers and Transcription Factor-Binding Sites in Chronic Myelomonocytic Leukemia.Cancer Res. 2015 Jul 15;75(14):2833-43. doi: 10.1158/0008-5472.CAN-14-0739. Epub 2015 May 13. Cancer Res. 2015. PMID: 25972343 Free PMC article. Clinical Trial.
-
Effects of TET2 mutations on DNA methylation in chronic myelomonocytic leukemia.Epigenetics. 2012 Feb;7(2):201-7. doi: 10.4161/epi.7.2.19015. Epigenetics. 2012. PMID: 22395470 Free PMC article.
-
TET2: A cornerstone in normal and malignant hematopoiesis.Cancer Sci. 2021 Jan;112(1):31-40. doi: 10.1111/cas.14688. Epub 2020 Nov 18. Cancer Sci. 2021. PMID: 33048426 Free PMC article. Review.
-
Dysregulation of TET2 in hematologic malignancies.Int J Hematol. 2017 Jan;105(1):17-22. doi: 10.1007/s12185-016-2122-z. Epub 2016 Nov 15. Int J Hematol. 2017. PMID: 27848178 Review.
Cited by
-
Validation of DNA methylation profiling in formalin-fixed paraffin-embedded samples using the Infinium HumanMethylation450 Microarray.Epigenetics. 2014 Jun;9(6):829-33. doi: 10.4161/epi.28790. Epub 2014 Apr 14. Epigenetics. 2014. PMID: 24732293 Free PMC article.
-
Plasma Circulating Tumor DNA in Patients with Primary Central Nervous System Lymphoma.Cancer Res Treat. 2022 Apr;54(2):597-612. doi: 10.4143/crt.2021.752. Epub 2021 Jul 23. Cancer Res Treat. 2022. PMID: 34325497 Free PMC article.
-
The interplay between DNA and histone methylation: molecular mechanisms and disease implications.EMBO Rep. 2021 May 5;22(5):e51803. doi: 10.15252/embr.202051803. Epub 2021 Apr 12. EMBO Rep. 2021. PMID: 33844406 Free PMC article. Review.
-
Physiological and pathological implications of 5-hydroxymethylcytosine in diseases.Oncotarget. 2016 Jul 26;7(30):48813-48831. doi: 10.18632/oncotarget.9281. Oncotarget. 2016. PMID: 27183914 Free PMC article. Review.
-
Non-catalytic Roles of Tet2 Are Essential to Regulate Hematopoietic Stem and Progenitor Cell Homeostasis.Cell Rep. 2019 Sep 3;28(10):2480-2490.e4. doi: 10.1016/j.celrep.2019.07.094. Cell Rep. 2019. PMID: 31484061 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
