

Glia and pain: is chronic pain a gliopathy?
- PMID: 23792284
- PMCID: PMC3858488
- DOI: 10.1016/j.pain.2013.06.022
Glia and pain: is chronic pain a gliopathy?
Abstract
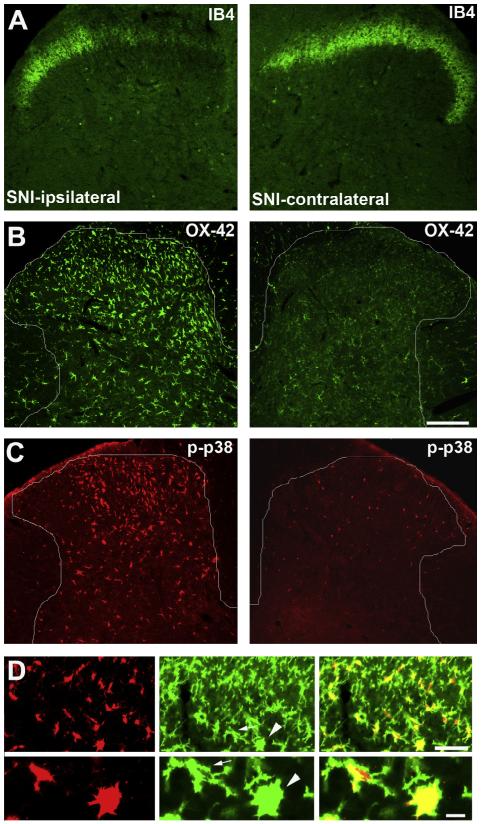
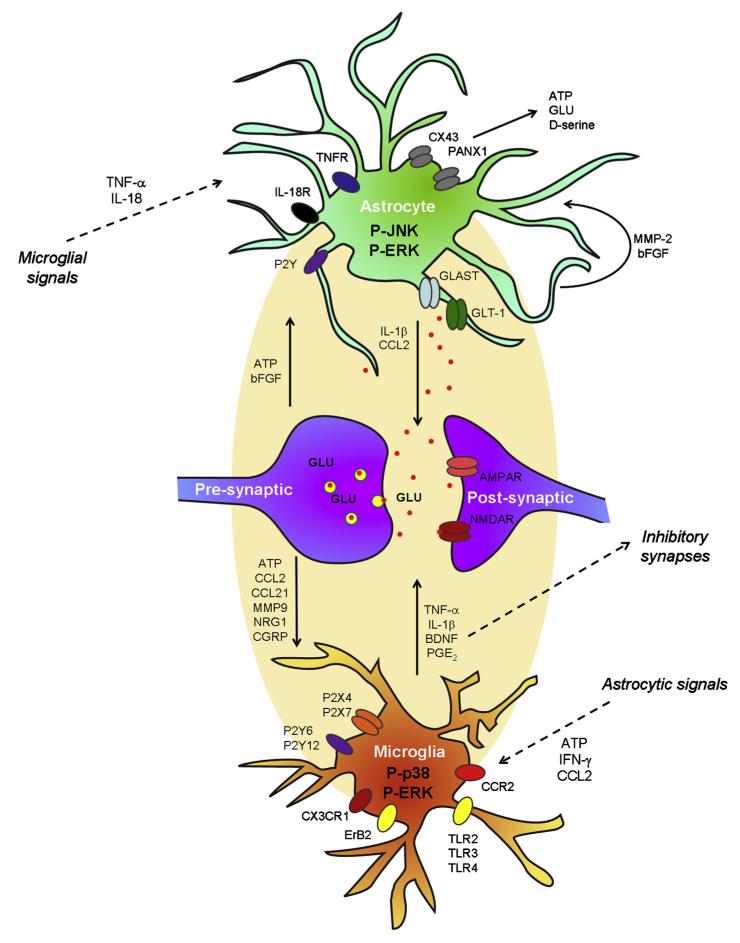
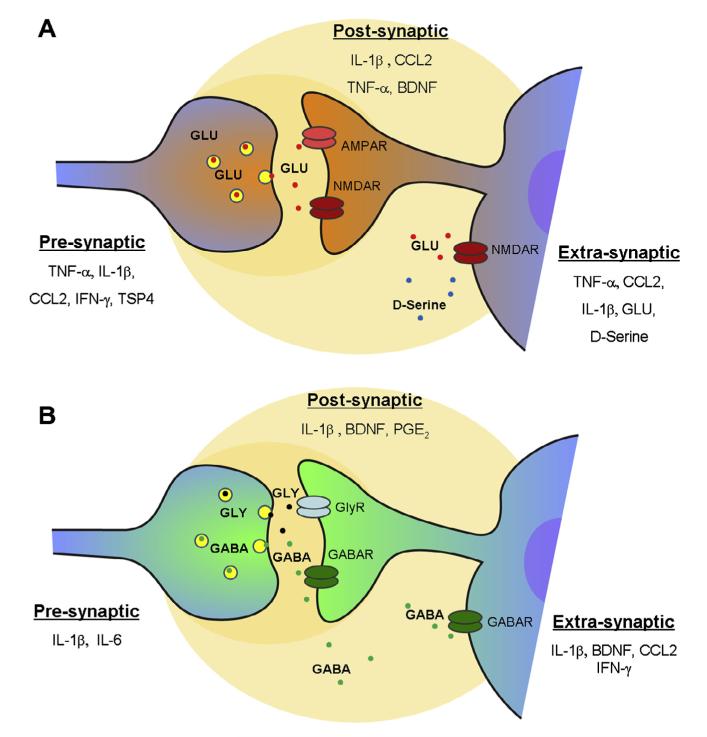
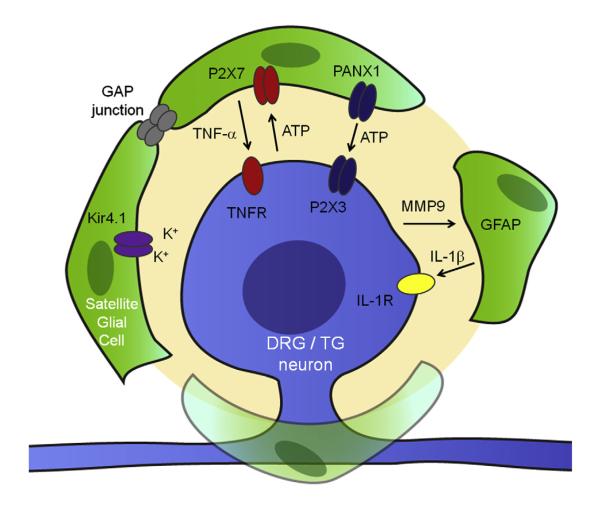
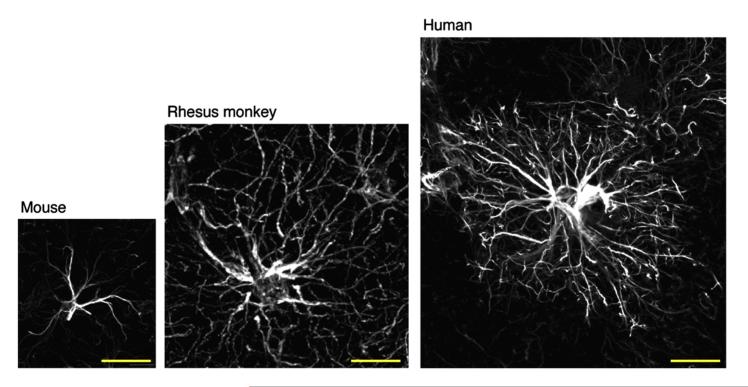
Activation of glial cells and neuro-glial interactions are emerging as key mechanisms underlying chronic pain. Accumulating evidence has implicated 3 types of glial cells in the development and maintenance of chronic pain: microglia and astrocytes of the central nervous system (CNS), and satellite glial cells of the dorsal root and trigeminal ganglia. Painful syndromes are associated with different glial activation states: (1) glial reaction (ie, upregulation of glial markers such as IBA1 and glial fibrillary acidic protein (GFAP) and/or morphological changes, including hypertrophy, proliferation, and modifications of glial networks); (2) phosphorylation of mitogen-activated protein kinase signaling pathways; (3) upregulation of adenosine triphosphate and chemokine receptors and hemichannels and downregulation of glutamate transporters; and (4) synthesis and release of glial mediators (eg, cytokines, chemokines, growth factors, and proteases) to the extracellular space. Although widely detected in chronic pain resulting from nerve trauma, inflammation, cancer, and chemotherapy in rodents, and more recently, human immunodeficiency virus-associated neuropathy in human beings, glial reaction (activation state 1) is not thought to mediate pain sensitivity directly. Instead, activation states 2 to 4 have been demonstrated to enhance pain sensitivity via a number of synergistic neuro-glial interactions. Glial mediators have been shown to powerfully modulate excitatory and inhibitory synaptic transmission at presynaptic, postsynaptic, and extrasynaptic sites. Glial activation also occurs in acute pain conditions, and acute opioid treatment activates peripheral glia to mask opioid analgesia. Thus, chronic pain could be a result of "gliopathy," that is, dysregulation of glial functions in the central and peripheral nervous system. In this review, we provide an update on recent advances and discuss remaining questions.
Keywords: ATP receptors; Astrocytes; Chemokines; Cytokines; Human; Microglia; Rodents; Satellite glial cells; Spinal cord.
Copyright © 2013 International Association for the Study of Pain. Published by Elsevier B.V. All rights reserved.
Figures

Similar articles
-
Spatial and temporal activation of spinal glial cells: role of gliopathy in central neuropathic pain following spinal cord injury in rats.Exp Neurol. 2012 Apr;234(2):362-72. doi: 10.1016/j.expneurol.2011.10.010. Epub 2011 Oct 21. Exp Neurol. 2012. PMID: 22036747 Free PMC article. Review.
-
Perspectives in Pain Research 2014: Neuroinflammation and glial cell activation: The cause of transition from acute to chronic pain?Scand J Pain. 2015 Jan 1;6(1):3-6. doi: 10.1016/j.sjpain.2014.10.002. Scand J Pain. 2015. PMID: 29911589
-
Influence of the vanilloid receptor TRPV1 on the activation of spinal cord glia in mouse models of pain.Exp Neurol. 2009 Dec;220(2):383-90. doi: 10.1016/j.expneurol.2009.09.030. Epub 2009 Oct 6. Exp Neurol. 2009. PMID: 19815011 Free PMC article.
-
The contribution of spinal glial cells to chronic pain behaviour in the monosodium iodoacetate model of osteoarthritic pain.Mol Pain. 2011 Nov 17;7:88. doi: 10.1186/1744-8069-7-88. Mol Pain. 2011. PMID: 22093915 Free PMC article.
-
Central Nervous System Targets: Glial Cell Mechanisms in Chronic Pain.Neurotherapeutics. 2020 Jul;17(3):846-860. doi: 10.1007/s13311-020-00905-7. Neurotherapeutics. 2020. PMID: 32820378 Free PMC article. Review.
Cited by
-
Ventrolateral Periaqueductal Gray Astrocytes Regulate Nociceptive Sensation and Emotional Motivation in Diabetic Neuropathic Pain.J Neurosci. 2022 Oct 26;42(43):8184-8199. doi: 10.1523/JNEUROSCI.0920-22.2022. Epub 2022 Sep 15. J Neurosci. 2022. PMID: 36109166 Free PMC article.
-
Involvement of CX3CL1/CX3CR1 signaling in spinal long term potentiation.PLoS One. 2015 Mar 13;10(3):e0118842. doi: 10.1371/journal.pone.0118842. eCollection 2015. PLoS One. 2015. PMID: 25768734 Free PMC article.
-
Macrophage-Colony Stimulating Factor Derived from Injured Primary Afferent Induces Proliferation of Spinal Microglia and Neuropathic Pain in Rats.PLoS One. 2016 Apr 12;11(4):e0153375. doi: 10.1371/journal.pone.0153375. eCollection 2016. PLoS One. 2016. PMID: 27071004 Free PMC article.
-
Glia and Orofacial Pain: Progress and Future Directions.Int J Mol Sci. 2021 May 19;22(10):5345. doi: 10.3390/ijms22105345. Int J Mol Sci. 2021. PMID: 34069553 Free PMC article. Review.
-
p38 phosphorylation in medullary microglia mediates ectopic orofacial inflammatory pain in rats.Mol Pain. 2015 Aug 12;11:48. doi: 10.1186/s12990-015-0053-y. Mol Pain. 2015. PMID: 26260484 Free PMC article.
References
-
- Adrian EK, Jr, Williams MG, George FC. Fine structure of reactive cells in injured nervous tissue labeled with 3H-thymidine injected before injury. J Comp Neurol. 1978;180:815–39. - PubMed
-
- Agulhon C, Fiacco TA, McCarthy KD. Hippocampal short-and long-term plasticity are not modulated by astrocyte Ca2+signaling. Science. 2010;327:1250–4. - PubMed
-
- Ahmadi S, Lippross S, Neuhuber WL, Zeilhofer HU. PGE(2) selectively blocks inhibitory glycinergic neurotransmission onto rat superficial dorsal horn neurons. Nat Neurosci. 2002;5:34–40. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
