

Idiopathic inflammatory myopathies: pathogenic mechanisms of muscle weakness
- PMID: 23758833
- PMCID: PMC3681571
- DOI: 10.1186/2044-5040-3-13
Idiopathic inflammatory myopathies: pathogenic mechanisms of muscle weakness
Abstract
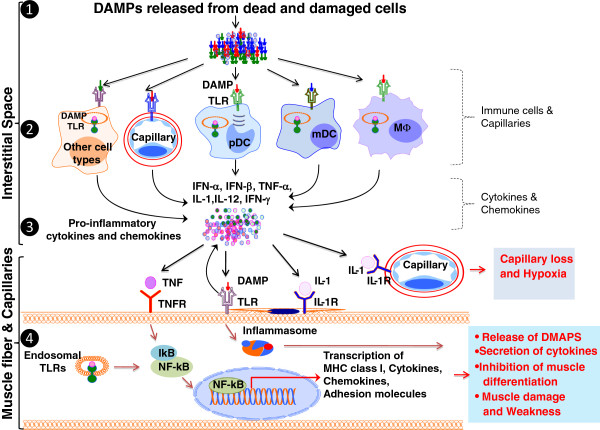
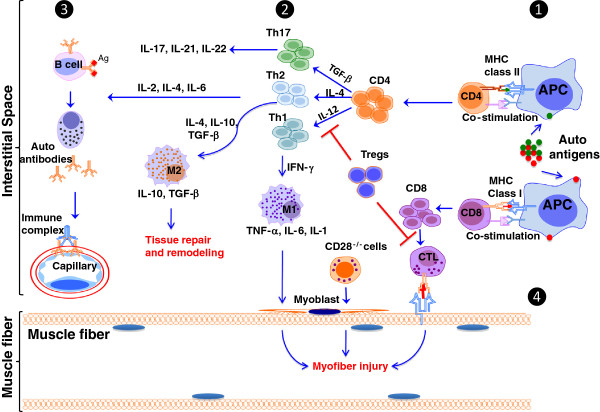
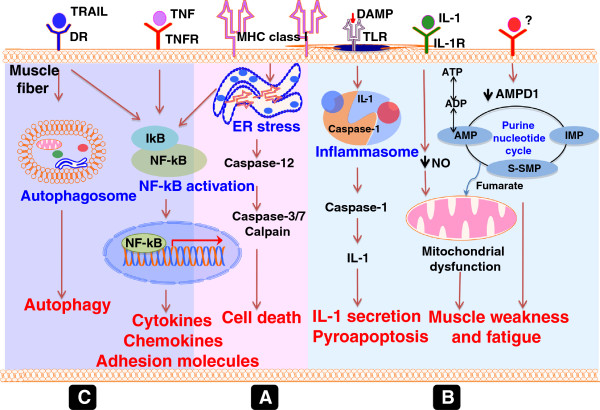
Idiopathic inflammatory myopathies (IIMs) are a heterogenous group of complex muscle diseases of unknown etiology. These diseases are characterized by progressive muscle weakness and damage, together with involvement of other organ systems. It is generally believed that the autoimmune response (autoreactive lymphocytes and autoantibodies) to skeletal muscle-derived antigens is responsible for the muscle fiber damage and muscle weakness in this group of disorders. Therefore, most of the current therapeutic strategies are directed at either suppressing or modifying immune cell activity. Recent studies have indicated that the underlying mechanisms that mediate muscle damage and dysfunction are multiple and complex. Emerging evidence indicates that not only autoimmune responses but also innate immune and non-immune metabolic pathways contribute to disease pathogenesis. However, the relative contributions of each of these mechanisms to disease pathogenesis are currently unknown. Here we discuss some of these complex pathways, their inter-relationships and their relation to muscle damage in myositis. Understanding the relative contributions of each of these pathways to disease pathogenesis would help us to identify suitable drug targets to alleviate muscle damage and also improve muscle weakness and quality of life for patients suffering from these debilitating muscle diseases.
Figures
Similar articles
-
Role of non-immune mechanisms of muscle damage in idiopathic inflammatory myopathies.Arthritis Res Ther. 2012 Apr 27;14(2):209. doi: 10.1186/ar3791. Arthritis Res Ther. 2012. PMID: 22546362 Free PMC article. Review.
-
In the idiopathic inflammatory myopathies (IIM), do reactive oxygen species (ROS) contribute to muscle weakness?Ann Rheum Dis. 2015 Jul;74(7):1340-6. doi: 10.1136/annrheumdis-2014-207172. Epub 2015 Apr 28. Ann Rheum Dis. 2015. PMID: 26063809 Review.
-
Muscle fiber necroptosis in pathophysiology of idiopathic inflammatory myopathies and its potential as target of novel treatment strategy.Front Immunol. 2023 Jul 7;14:1191815. doi: 10.3389/fimmu.2023.1191815. eCollection 2023. Front Immunol. 2023. PMID: 37483632 Free PMC article. Review.
-
[Idiopathic inflammatory myopathies from the viewpoint of a neurologist].Brain Nerve. 2013 Nov;65(11):1269-74. Brain Nerve. 2013. PMID: 24200604 Review. Japanese.
-
The altered metabolism profile in pathogenesis of idiopathic inflammatory myopathies.Semin Arthritis Rheum. 2020 Aug;50(4):627-635. doi: 10.1016/j.semarthrit.2020.05.008. Epub 2020 May 25. Semin Arthritis Rheum. 2020. PMID: 32502727 Review.
Cited by
-
Clinico pathological study of adult dermatomyositis: Importance of muscle histology in the diagnosis.Ann Indian Acad Neurol. 2015 Apr-Jun;18(2):194-9. doi: 10.4103/0972-2327.150603. Ann Indian Acad Neurol. 2015. PMID: 26019418 Free PMC article.
-
Ccn6 Is Required for Mitochondrial Integrity and Skeletal Muscle Function in Zebrafish.Front Cell Dev Biol. 2021 Feb 11;9:627409. doi: 10.3389/fcell.2021.627409. eCollection 2021. Front Cell Dev Biol. 2021. PMID: 33644064 Free PMC article.
-
Muscle fibrosis as a prognostic biomarker in facioscapulohumeral muscular dystrophy: a retrospective cohort study.Acta Neuropathol Commun. 2023 Oct 17;11(1):165. doi: 10.1186/s40478-023-01660-4. Acta Neuropathol Commun. 2023. PMID: 37849014 Free PMC article.
-
Proteomic profiling of sporadic late-onset nemaline myopathy.Ann Clin Transl Neurol. 2022 Mar;9(3):391-402. doi: 10.1002/acn3.51527. Epub 2022 Feb 20. Ann Clin Transl Neurol. 2022. PMID: 35187860 Free PMC article.
-
TLR expression in peripheral monocyte subsets of patients with idiopathic inflammatory myopathies: association with clinical and immunological features.J Transl Med. 2020 Mar 12;18(1):125. doi: 10.1186/s12967-020-02290-3. J Transl Med. 2020. PMID: 32164729 Free PMC article.
References
-
- Zong M, Bruton JD, Grundtman C, Yang H, Li JH, Alexanderson H, Palmblad K, Andersson U, Harris HE, Lundberg IE, Westerblad H. TLR4 as receptor for HMGB1 induced muscle dysfunction in myositis. Ann Rheum Dis. 2012. Epub ahead of print. - PubMed
-
- Alger HM, Raben N, Pistilli E, Francia DL, Rawat R, Getnet D, Ghimbovschi S, Chen YW, Lundberg IE, Nagaraju K. The role of TRAIL in mediating autophagy in myositis skeletal muscle: a potential nonimmune mechanism of muscle damage. Arthritis Rheum. 2011;63:3448–3457. doi: 10.1002/art.30530. - DOI - PMC - PubMed
-
- Grundtman C, Bruton J, Yamada T, Ostberg T, Pisetsky DS, Harris HE, Andersson U, Lundberg IE, Westerblad H. Effects of HMGB1 on in vitro responses of isolated muscle fibers and functional aspects in skeletal muscles of idiopathic inflammatory myopathies. FASEB J. 2010;24:570–578. doi: 10.1096/fj.09-144782. - DOI - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
