

Dissecting Major Signaling Pathways throughout the Development of Prostate Cancer
- PMID: 23738079
- PMCID: PMC3657461
- DOI: 10.1155/2013/920612
Dissecting Major Signaling Pathways throughout the Development of Prostate Cancer
Abstract
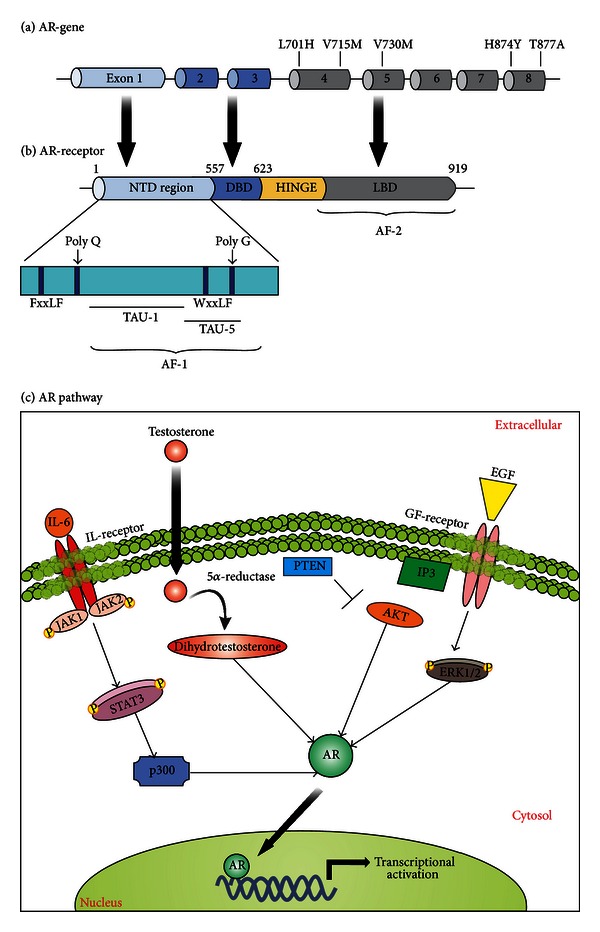
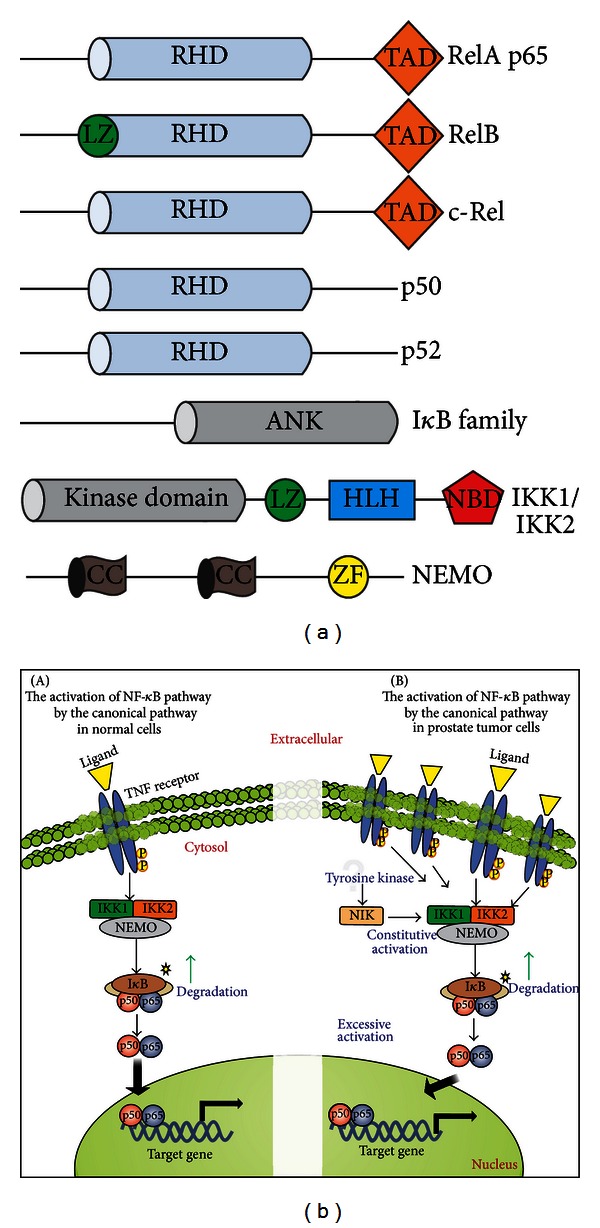
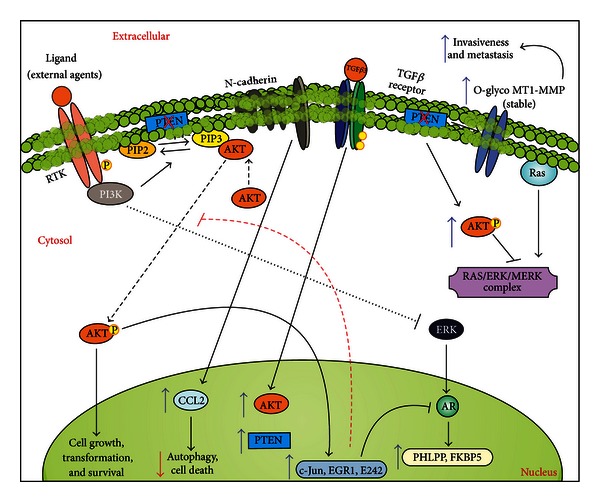
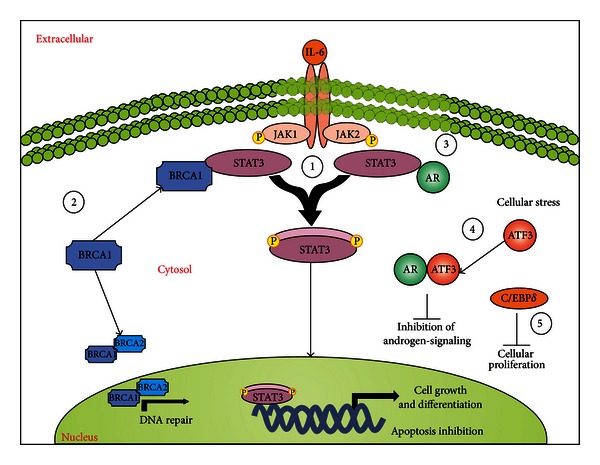
Prostate cancer (PCa) is one of the most common malignancies found in males. The development of PCa involves several mutations in prostate epithelial cells, usually linked to developmental changes, such as enhanced resistance to apoptotic death, constitutive proliferation, and, in some cases, to differentiation into an androgen deprivation-resistant phenotype, leading to the appearance of castration-resistant PCa (CRPCa), which leads to a poor prognosis in patients. In this review, we summarize recent findings concerning the main deregulations into signaling pathways that will lead to the development of PCa and/or CRPCa. Key mutations in some pathway molecules are often linked to a higher prevalence of PCa, by directly affecting the respective cascade and, in some cases, by deregulating a cross-talk node or junction along the pathways. We also discuss the possible environmental and nonenvironmental inducers for these mutations, as well as the potential therapeutic strategies targeting these signaling pathways. A better understanding of how some risk factors induce deregulation of these signaling pathways, as well as how these deregulated pathways affect the development of PCa and CRPCa, will further help in the development of new treatments and prevention strategies for this disease.
Figures
Similar articles
-
From pathogenesis to prevention of castration resistant prostate cancer.Prostate. 2010 Jan 1;70(1):100-12. doi: 10.1002/pros.21042. Prostate. 2010. PMID: 19760632 Review.
-
Wnt/Beta-Catenin Signaling and Prostate Cancer Therapy Resistance.Adv Exp Med Biol. 2019;1210:351-378. doi: 10.1007/978-3-030-32656-2_16. Adv Exp Med Biol. 2019. PMID: 31900917 Review.
-
Targeting treatment options for castration-resistant prostate cancer.Am J Clin Exp Urol. 2021 Feb 15;9(1):101-120. eCollection 2021. Am J Clin Exp Urol. 2021. PMID: 33816699 Free PMC article. Review.
-
A Novel Small Molecule Inhibits Tumor Growth and Synergizes Effects of Enzalutamide on Prostate Cancer.J Pharmacol Exp Ther. 2019 Dec;371(3):703-712. doi: 10.1124/jpet.119.261040. Epub 2019 Oct 3. J Pharmacol Exp Ther. 2019. PMID: 31582422 Free PMC article.
-
NF-κB signaling promotes castration-resistant prostate cancer initiation and progression.Pharmacol Ther. 2020 Jul;211:107538. doi: 10.1016/j.pharmthera.2020.107538. Epub 2020 Mar 19. Pharmacol Ther. 2020. PMID: 32201312 Review.
Cited by
-
Prostate cancer health disparities: An immuno-biological perspective.Cancer Lett. 2018 Feb 1;414:153-165. doi: 10.1016/j.canlet.2017.11.011. Epub 2017 Nov 15. Cancer Lett. 2018. PMID: 29154974 Free PMC article.
-
Lycopene inhibits the cell proliferation and invasion of human head and neck squamous cell carcinoma.Mol Med Rep. 2016 Oct;14(4):2953-8. doi: 10.3892/mmr.2016.5597. Epub 2016 Aug 8. Mol Med Rep. 2016. PMID: 27510325 Free PMC article.
-
WMAXC: a weighted maximum clique method for identifying condition-specific sub-network.PLoS One. 2014 Aug 22;9(8):e104993. doi: 10.1371/journal.pone.0104993. eCollection 2014. PLoS One. 2014. PMID: 25148538 Free PMC article.
-
Identification of shared and unique susceptibility pathways among cancers of the lung, breast, and prostate from genome-wide association studies and tissue-specific protein interactions.Hum Mol Genet. 2015 Dec 20;24(25):7406-20. doi: 10.1093/hmg/ddv440. Epub 2015 Oct 19. Hum Mol Genet. 2015. PMID: 26483192 Free PMC article.
-
Combined genetic and epigenetic interferences with interferon signaling expose prostate cancer cells to viral infection.Oncotarget. 2016 Aug 9;7(32):52115-52134. doi: 10.18632/oncotarget.10313. Oncotarget. 2016. PMID: 27366948 Free PMC article.
References
-
- Verras M, Sun Z. Roles and regulation of Wnt signaling and β-catenin in prostate cancer. Cancer Letters. 2006;237(1):22–32. - PubMed
-
- Mullins JK, Loeb S. Environmental exposures and prostate cancer. Urologic Oncology. 2012;30:216–219. - PubMed
-
- Siegel R, DeSantis C, Virgo K, et al. Cancer treatment and survivorship statistics, 2012. CA: A Cancer Journal for Clinicians. 2012;62:220–241. - PubMed
-
- Center MM, Jemal A, Lortet-Tieulent J, et al. International variation in prostate cancer incidence and mortality rates. European Urology. 2012;61:1079–1092. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
