

Mitochondrial damage revealed by immunoselection for ALS-linked misfolded SOD1
- PMID: 23736301
- PMCID: PMC5052069
- DOI: 10.1093/hmg/ddt249
Mitochondrial damage revealed by immunoselection for ALS-linked misfolded SOD1
Abstract
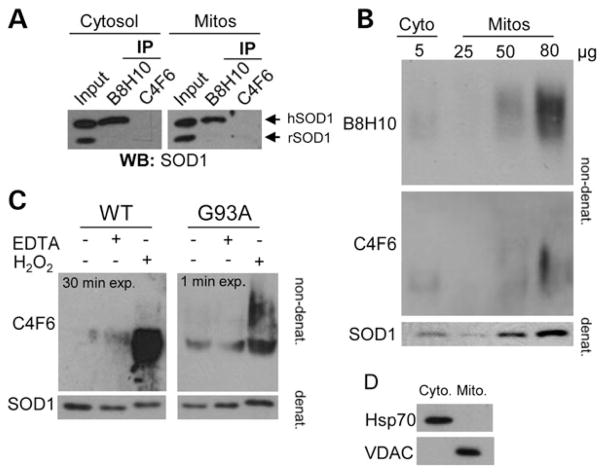
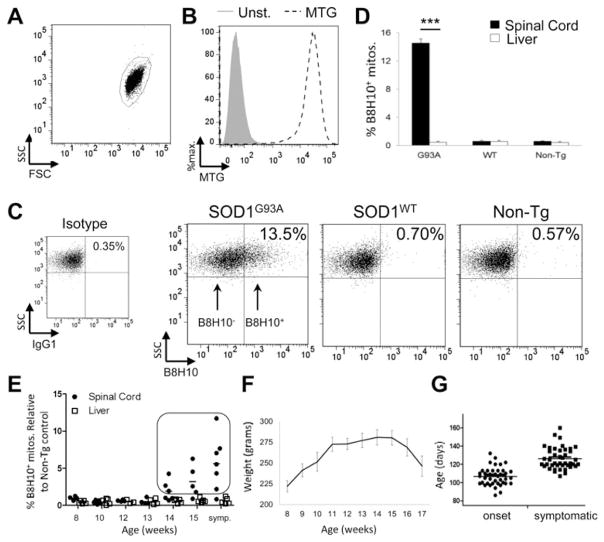
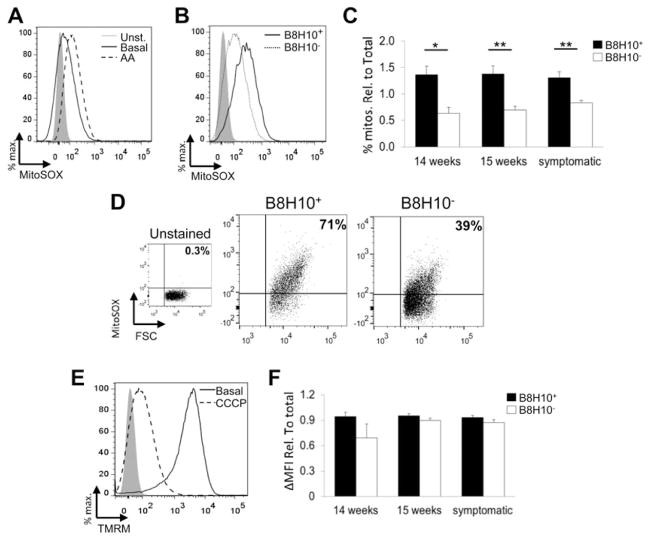
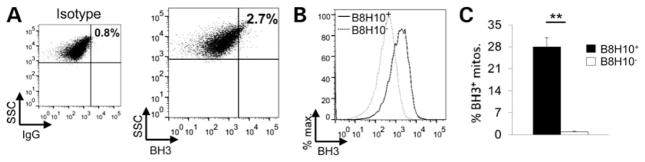
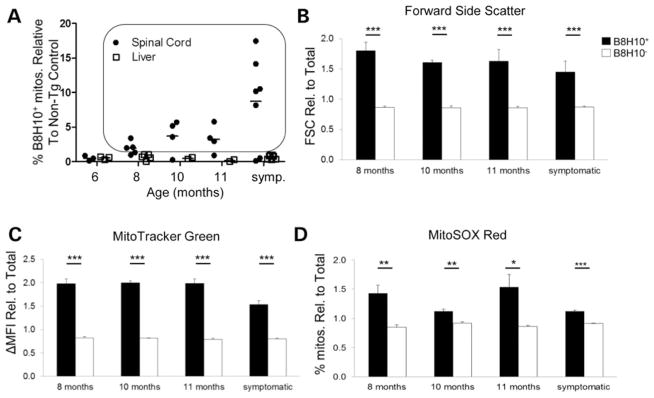
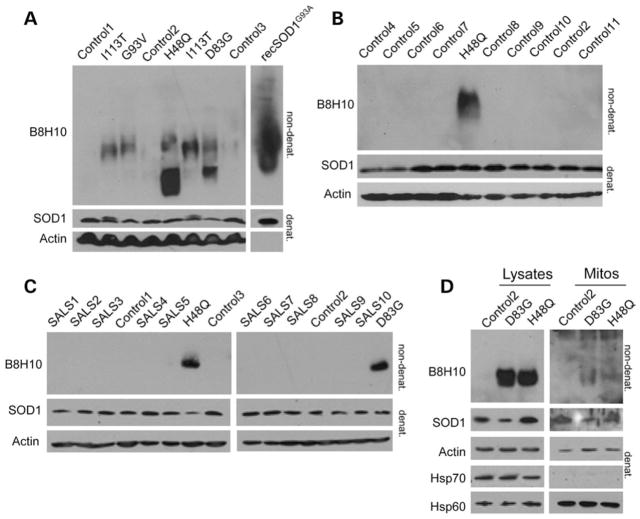
Mutant superoxide dismutase 1 (SOD1) selectively associates with spinal cord mitochondria in rodent models of SOD1-mediated amyotrophic lateral sclerosis. A portion of mutant SOD1 exists in a non-native/misfolded conformation that is selectively recognized by conformational antibodies. Misfolded SOD1 is common to all mutant SOD1 models, is uniquely found in areas affected by the disease and is considered to mediate toxicity. We report that misfolded SOD1 recognized by the antibody B8H10 is present in greater abundance in mitochondrial fractions of SOD1(G93A) rat spinal cords compared with oxidized SOD1, as recognized by the C4F6 antibody. Using a novel flow cytometric assay, we detect an age-dependent deposition of B8H10-reactive SOD1 on spinal cord mitochondria from both SOD1(G93A) rats and SOD1(G37R) mice. Mitochondrial damage, including increased mitochondrial volume, excess superoxide production and increased exposure of the toxic BH3 domain of Bcl-2, tracks positively with the presence of misfolded SOD1. Lastly, B8H10 reactive misfolded SOD1 is present in the lysates and mitochondrial fractions of lymphoblasts derived from ALS patients carrying SOD1 mutations, but not in controls. Together, these results highlight misfolded SOD1 as common to two ALS rodent animal models and familial ALS patient lymphoblasts with four different SOD1 mutations. Studies in the animal models point to a role for misfolded SOD1 in mitochondrial dysfunction in ALS pathogenesis.
Conflict of interest statement
statement. None declared.
Figures


Similar articles
-
ALS-linked misfolded SOD1 species have divergent impacts on mitochondria.Acta Neuropathol Commun. 2016 Apr 27;4(1):43. doi: 10.1186/s40478-016-0313-8. Acta Neuropathol Commun. 2016. PMID: 27121871 Free PMC article.
-
Identification of a misfolded region in superoxide dismutase 1 that is exposed in amyotrophic lateral sclerosis.J Biol Chem. 2014 Oct 10;289(41):28527-38. doi: 10.1074/jbc.M114.581801. Epub 2014 Aug 27. J Biol Chem. 2014. PMID: 25164820 Free PMC article.
-
Proteins that bind to misfolded mutant superoxide dismutase-1 in spinal cords from transgenic amyotrophic lateral sclerosis (ALS) model mice.J Biol Chem. 2011 Jun 10;286(23):20130-6. doi: 10.1074/jbc.M111.218842. Epub 2011 Apr 14. J Biol Chem. 2011. PMID: 21493711 Free PMC article.
-
Misfolded SOD1 and ALS: zeroing in on mitochondria.Amyotroph Lateral Scler. 2012 Jun;13(4):333-40. doi: 10.3109/17482968.2012.648645. Epub 2012 Apr 3. Amyotroph Lateral Scler. 2012. PMID: 22471903 Review.
-
Oxidized/misfolded superoxide dismutase-1: the cause of all amyotrophic lateral sclerosis?Ann Neurol. 2007 Dec;62(6):553-9. doi: 10.1002/ana.21319. Ann Neurol. 2007. PMID: 18074357 Review.
Cited by
-
Endogenous macrophage migration inhibitory factor reduces the accumulation and toxicity of misfolded SOD1 in a mouse model of ALS.Proc Natl Acad Sci U S A. 2016 Sep 6;113(36):10198-203. doi: 10.1073/pnas.1604600113. Epub 2016 Aug 22. Proc Natl Acad Sci U S A. 2016. PMID: 27551074 Free PMC article.
-
Stage-dependent remodeling of projections to motor cortex in ALS mouse model revealed by a new variant retrograde-AAV9.Elife. 2018 Aug 23;7:e36892. doi: 10.7554/eLife.36892. Elife. 2018. PMID: 30136928 Free PMC article.
-
Morpholino-mediated SOD1 reduction ameliorates an amyotrophic lateral sclerosis disease phenotype.Sci Rep. 2016 Feb 16;6:21301. doi: 10.1038/srep21301. Sci Rep. 2016. PMID: 26878886 Free PMC article.
-
Regulation of mitophagy by the ubiquitin pathway in neurodegenerative diseases.Exp Biol Med (Maywood). 2018 Mar;243(6):554-562. doi: 10.1177/1535370217752351. Epub 2018 Jan 9. Exp Biol Med (Maywood). 2018. PMID: 29316798 Free PMC article. Review.
-
Therapeutic rAAVrh10 Mediated SOD1 Silencing in Adult SOD1(G93A) Mice and Nonhuman Primates.Hum Gene Ther. 2016 Jan;27(1):19-31. doi: 10.1089/hum.2015.122. Hum Gene Ther. 2016. PMID: 26710998 Free PMC article.
References
-
- Boillee S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006;52:1–21. - PubMed
-
- Sasaki S, Iwata M. Impairment of fast axonal transport in the proximal axons of anterior horn neurons in amyotrophic lateral sclerosis. Neurology. 1996;47:535–540. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
