

Regulated ADAM17-dependent EGF family ligand release by substrate-selecting signaling pathways
- PMID: 23720309
- PMCID: PMC3683718
- DOI: 10.1073/pnas.1307478110
Regulated ADAM17-dependent EGF family ligand release by substrate-selecting signaling pathways
Abstract
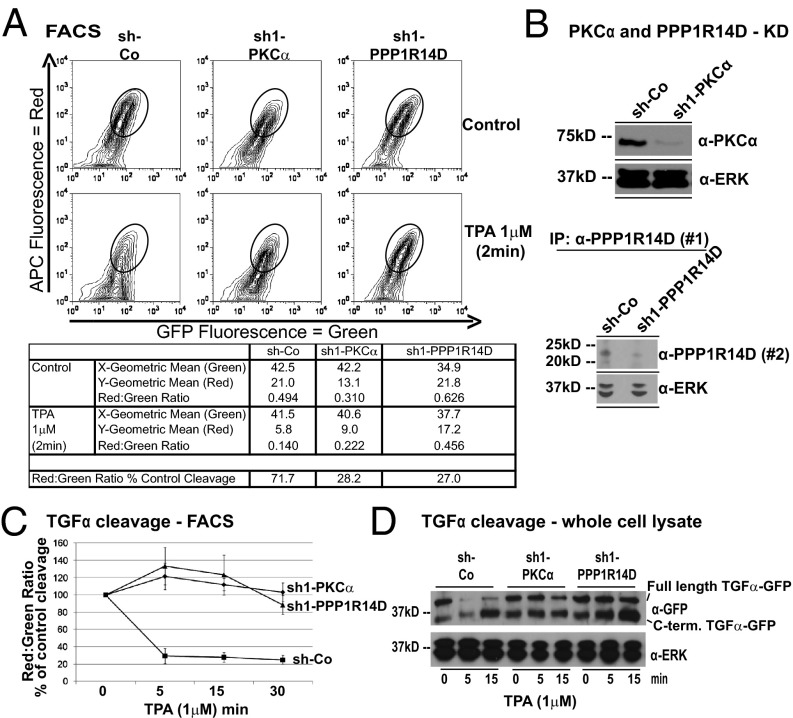
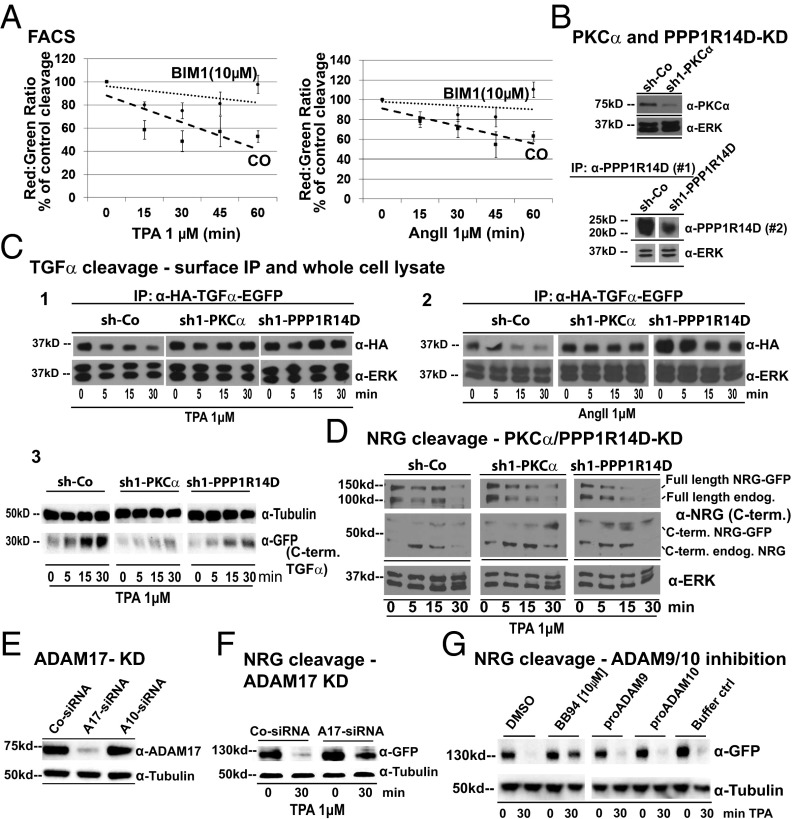
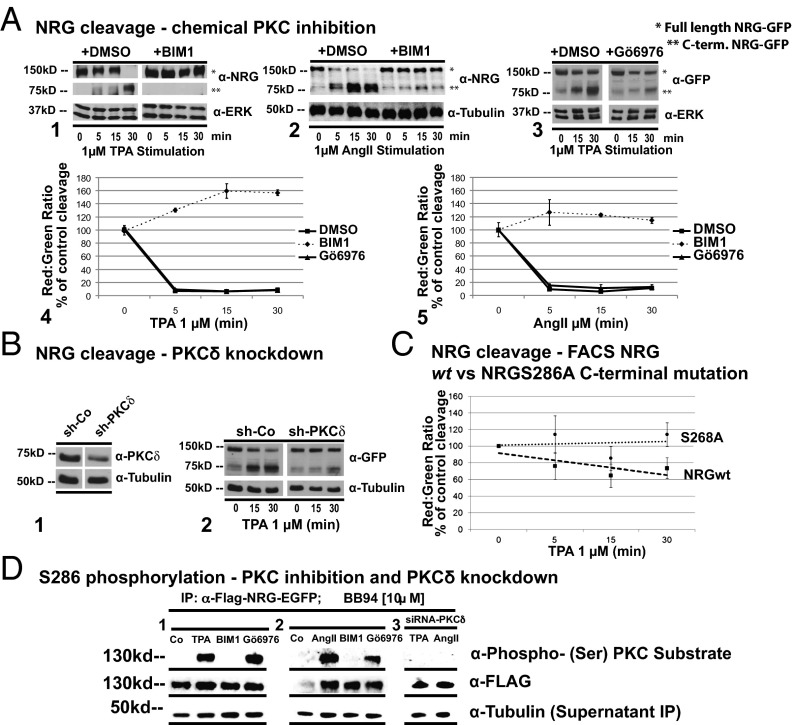
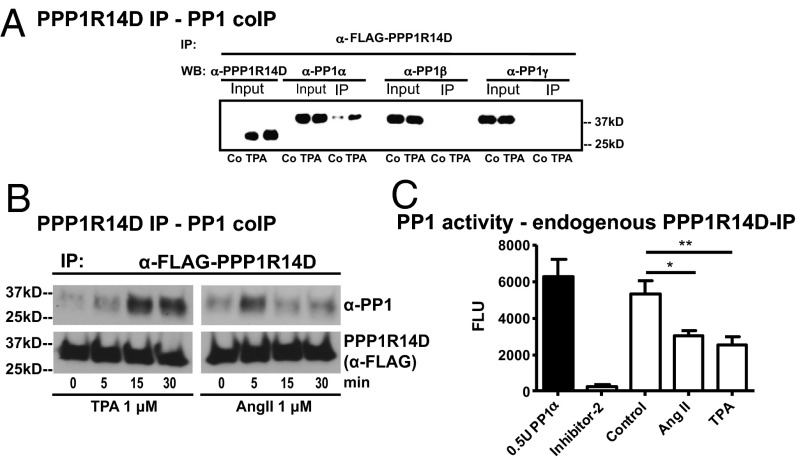
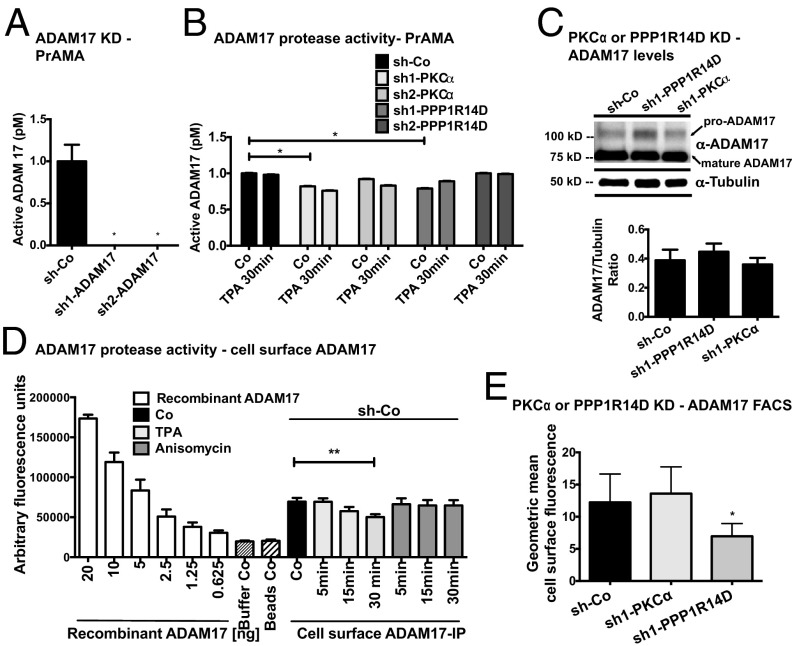
Ectodomain cleavage of cell-surface proteins by A disintegrin and metalloproteinases (ADAMs) is highly regulated, and its dysregulation has been linked to many diseases. ADAM10 and ADAM17 cleave most disease-relevant substrates. Broad-spectrum metalloprotease inhibitors have failed clinically, and targeting the cleavage of a specific substrate has remained impossible. It is therefore necessary to identify signaling intermediates that determine substrate specificity of cleavage. We show here that phorbol ester or angiotensin II-induced proteolytic release of EGF family members may not require a significant increase in ADAM17 protease activity. Rather, inducers activate a signaling pathway using PKC-α and the PKC-regulated protein phosphatase 1 inhibitor 14D that is required for ADAM17 cleavage of TGF-α, heparin-binding EGF, and amphiregulin. A second pathway involving PKC-δ is required for neuregulin (NRG) cleavage, and, indeed, PKC-δ phosphorylation of serine 286 in the NRG cytosolic domain is essential for induced NRG cleavage. Thus, signaling-mediated substrate selection is clearly distinct from regulation of enzyme activity, an important mechanism that offers itself for application in disease.
Keywords: epidermal growth factor receptor; transactivation.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Substrate selectivity of epidermal growth factor-receptor ligand sheddases and their regulation by phorbol esters and calcium influx.Mol Biol Cell. 2007 Jan;18(1):176-88. doi: 10.1091/mbc.e06-01-0014. Epub 2006 Nov 1. Mol Biol Cell. 2007. PMID: 17079736 Free PMC article.
-
Epidermal growth factor (EGF) ligand release by substrate-specific a disintegrin and metalloproteases (ADAMs) involves different protein kinase C (PKC) isoenzymes depending on the stimulus.J Biol Chem. 2011 May 20;286(20):17704-13. doi: 10.1074/jbc.M110.187823. Epub 2011 Mar 22. J Biol Chem. 2011. PMID: 21454702 Free PMC article.
-
Hormone-induced expression of tumor necrosis factor alpha-converting enzyme/A disintegrin and metalloprotease-17 impacts porcine cumulus cell oocyte complex expansion and meiotic maturation via ligand activation of the epidermal growth factor receptor.Endocrinology. 2007 Dec;148(12):6164-75. doi: 10.1210/en.2007-0195. Epub 2007 Sep 27. Endocrinology. 2007. PMID: 17901238
-
TACE/ADAM17 processing of EGFR ligands indicates a role as a physiological convertase.Ann N Y Acad Sci. 2003 May;995:22-38. doi: 10.1111/j.1749-6632.2003.tb03207.x. Ann N Y Acad Sci. 2003. PMID: 12814936 Review.
-
The ADAM17-amphiregulin-EGFR axis in mammary development and cancer.J Mammary Gland Biol Neoplasia. 2008 Jun;13(2):181-94. doi: 10.1007/s10911-008-9084-6. Epub 2008 May 10. J Mammary Gland Biol Neoplasia. 2008. PMID: 18470483 Free PMC article. Review.
Cited by
-
Polo-like kinase 2, a novel ADAM17 signaling component, regulates tumor necrosis factor α ectodomain shedding.J Biol Chem. 2014 Jan 31;289(5):3080-93. doi: 10.1074/jbc.M113.536847. Epub 2013 Dec 13. J Biol Chem. 2014. PMID: 24338472 Free PMC article.
-
Fine Tuning Cell Migration by a Disintegrin and Metalloproteinases.Mediators Inflamm. 2017;2017:9621724. doi: 10.1155/2017/9621724. Epub 2017 Feb 5. Mediators Inflamm. 2017. PMID: 28260841 Free PMC article. Review.
-
Recent Advances in ADAM17 Research: A Promising Target for Cancer and Inflammation.Mediators Inflamm. 2017;2017:9673537. doi: 10.1155/2017/9673537. Epub 2017 Nov 2. Mediators Inflamm. 2017. PMID: 29230082 Free PMC article. Review.
-
Contribution of ADAM17 and related ADAMs in cardiovascular diseases.Cell Mol Life Sci. 2021 May;78(9):4161-4187. doi: 10.1007/s00018-021-03779-w. Epub 2021 Feb 11. Cell Mol Life Sci. 2021. PMID: 33575814 Free PMC article. Review.
-
Protein Kinase C Inhibition Mediates Neuroblast Enrichment in Mechanical Brain Injuries.Front Cell Neurosci. 2018 Nov 27;12:462. doi: 10.3389/fncel.2018.00462. eCollection 2018. Front Cell Neurosci. 2018. PMID: 30542270 Free PMC article.
References
-
- Overall CM, Blobel CP. In search of partners: Linking extracellular proteases to substrates. Nat Rev Mol Cell Biol. 2007;8(3):245–257. - PubMed
-
- Murphy G. The ADAMs: Signalling scissors in the tumour microenvironment. Nat Rev Cancer. 2008;8(12):929–941. - PubMed
-
- Klein T, Bischoff R. Active metalloproteases of the A Disintegrin and Metalloprotease (ADAM) family: Biological function and structure. J Proteome Res. 2011;10(1):17–33. - PubMed
-
- Schneider MR, Wolf E. The epidermal growth factor receptor ligands at a glance. J Cell Physiol. 2009;218(3):460–466. - PubMed
-
- Higashiyama S, Nanba D, Nakayama H, Inoue H, Fukuda S. Ectodomain shedding and remnant peptide signalling of EGFRs and their ligands. J Biochem. 2011;150(1):15–22. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
