

Ab Initio structure prediction for Escherichia coli: towards genome-wide protein structure modeling and fold assignment
- PMID: 23719418
- PMCID: PMC3667494
- DOI: 10.1038/srep01895
Ab Initio structure prediction for Escherichia coli: towards genome-wide protein structure modeling and fold assignment
Abstract
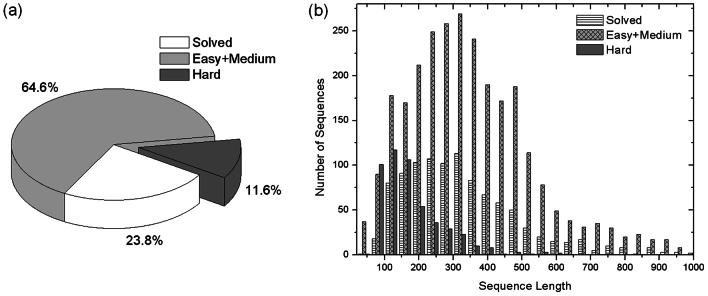
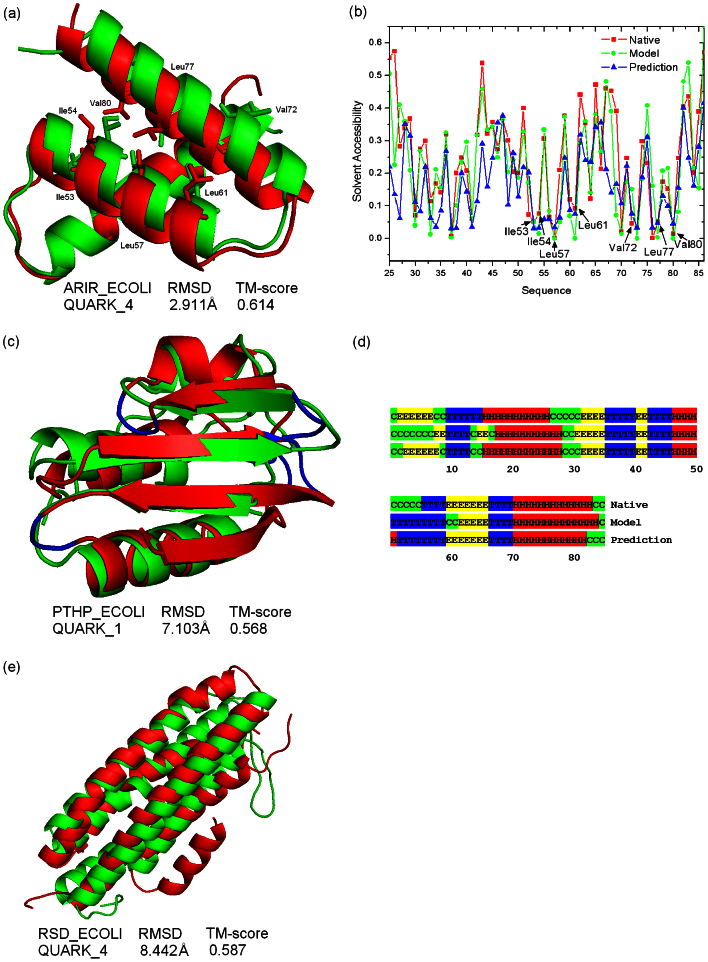
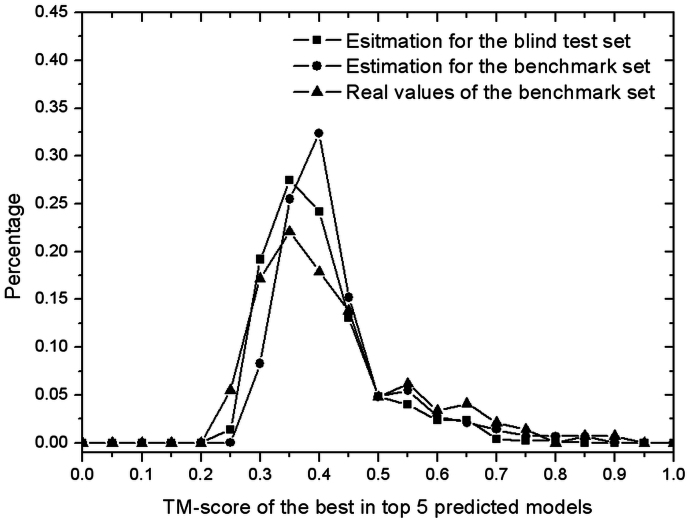
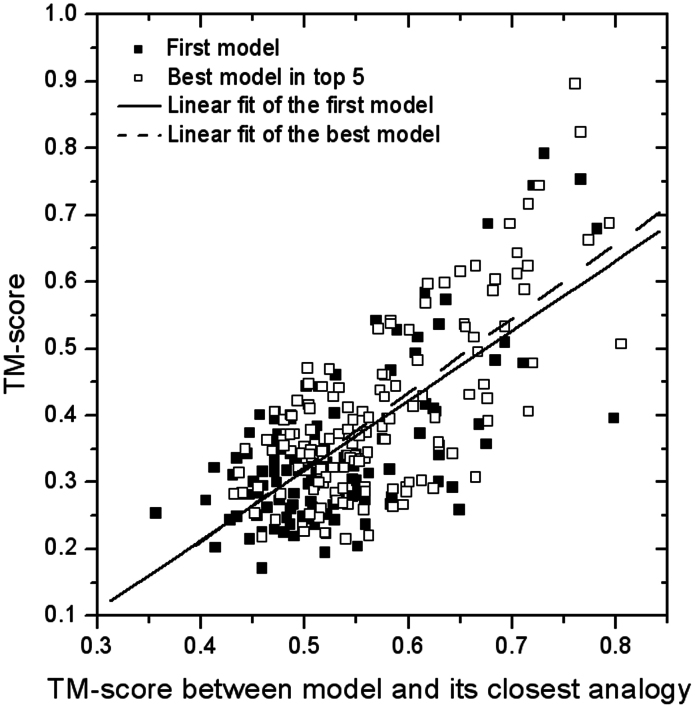
Genome-wide protein structure prediction and structure-based function annotation have been a long-term goal in molecular biology but not yet become possible due to difficulties in modeling distant-homology targets. We developed a hybrid pipeline combining ab initio folding and template-based modeling for genome-wide structure prediction applied to the Escherichia coli genome. The pipeline was tested on 43 known sequences, where QUARK-based ab initio folding simulation generated models with TM-score 17% higher than that by traditional comparative modeling methods. For 495 unknown hard sequences, 72 are predicted to have a correct fold (TM-score > 0.5) and 321 have a substantial portion of structure correctly modeled (TM-score > 0.35). 317 sequences can be reliably assigned to a SCOP fold family based on structural analogy to existing proteins in PDB. The presented results, as a case study of E. coli, represent promising progress towards genome-wide structure modeling and fold family assignment using state-of-the-art ab initio folding algorithms.
Figures

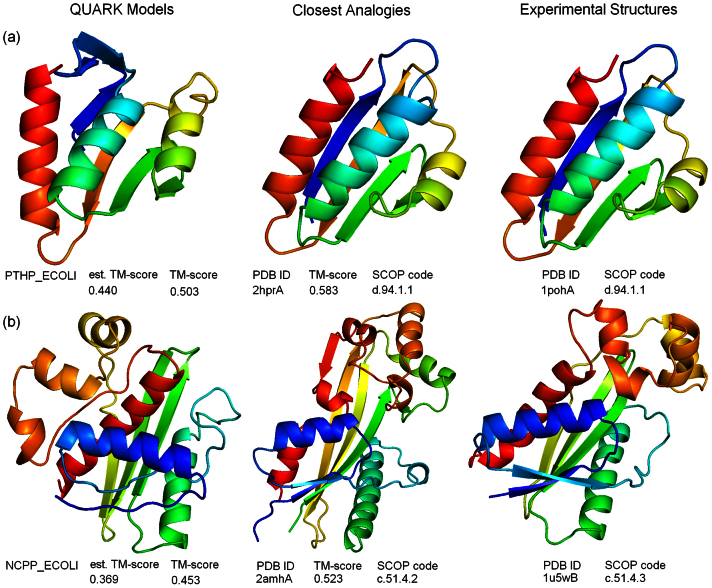
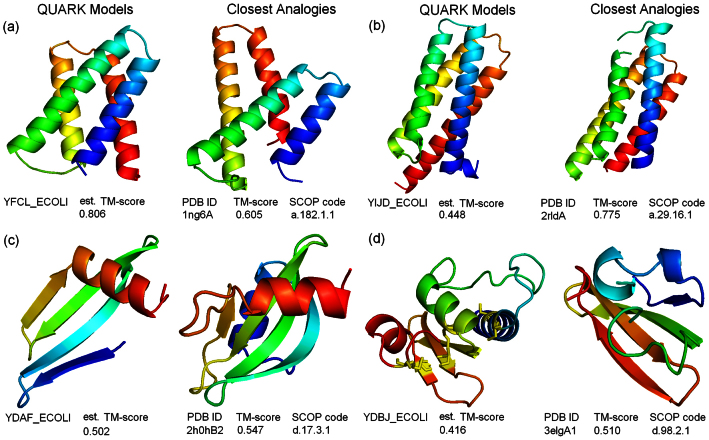
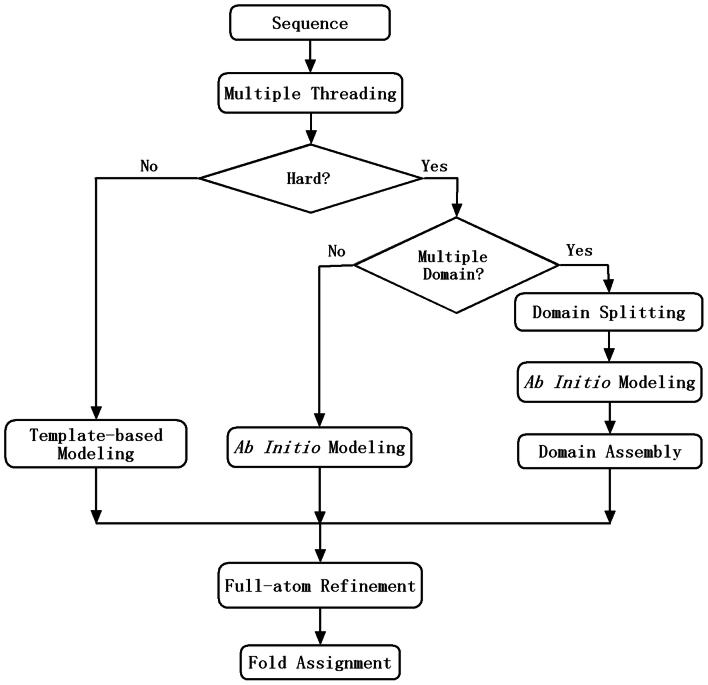
Similar articles
-
Integration of QUARK and I-TASSER for Ab Initio Protein Structure Prediction in CASP11.Proteins. 2016 Sep;84 Suppl 1(Suppl 1):76-86. doi: 10.1002/prot.24930. Epub 2015 Sep 23. Proteins. 2016. PMID: 26370505 Free PMC article.
-
Interplay of I-TASSER and QUARK for template-based and ab initio protein structure prediction in CASP10.Proteins. 2014 Feb;82 Suppl 2(0 2):175-87. doi: 10.1002/prot.24341. Epub 2013 Aug 31. Proteins. 2014. PMID: 23760925 Free PMC article.
-
Template-based protein structure prediction in CASP11 and retrospect of I-TASSER in the last decade.Proteins. 2016 Sep;84 Suppl 1(Suppl 1):233-46. doi: 10.1002/prot.24918. Epub 2015 Sep 18. Proteins. 2016. PMID: 26343917 Free PMC article.
-
Template-based protein modeling: recent methodological advances.Curr Top Med Chem. 2010;10(1):84-94. doi: 10.2174/156802610790232314. Curr Top Med Chem. 2010. PMID: 19929829 Free PMC article. Review.
-
Ab initio protein structure prediction: progress and prospects.Annu Rev Biophys Biomol Struct. 2001;30:173-89. doi: 10.1146/annurev.biophys.30.1.173. Annu Rev Biophys Biomol Struct. 2001. PMID: 11340057 Review.
Cited by
-
Molecular dynamics simulation reveals insights into the mechanism of unfolding by the A130T/V mutations within the MID1 zinc-binding Bbox1 domain.PLoS One. 2015 Apr 13;10(4):e0124377. doi: 10.1371/journal.pone.0124377. eCollection 2015. PLoS One. 2015. PMID: 25874572 Free PMC article.
-
Approaches to ab initio molecular replacement of α-helical transmembrane proteins.Acta Crystallogr D Struct Biol. 2017 Dec 1;73(Pt 12):985-996. doi: 10.1107/S2059798317016436. Epub 2017 Nov 22. Acta Crystallogr D Struct Biol. 2017. PMID: 29199978 Free PMC article.
-
Highly accurate protein structure prediction for the human proteome.Nature. 2021 Aug;596(7873):590-596. doi: 10.1038/s41586-021-03828-1. Epub 2021 Jul 22. Nature. 2021. PMID: 34293799 Free PMC article.
-
Selenoprotein N is an endoplasmic reticulum calcium sensor that links luminal calcium levels to a redox activity.Proc Natl Acad Sci U S A. 2020 Sep 1;117(35):21288-21298. doi: 10.1073/pnas.2003847117. Epub 2020 Aug 17. Proc Natl Acad Sci U S A. 2020. PMID: 32817544 Free PMC article.
-
A hybrid method for identification of structural domains.Sci Rep. 2014 Dec 15;4:7476. doi: 10.1038/srep07476. Sci Rep. 2014. PMID: 25503992 Free PMC article.
References
-
- Jensen O. N. Modification-specific proteomics: characterization of post-translational modifications by mass spectrometry. Curr Opin Chem Biol 8(1), 33–41 (2004). - PubMed
-
- Levitt M. & Warshel A. Computer-Simulation of Protein Folding. Nature 253(5494), 694–698 (1975). - PubMed
-
- Mccammon J. A., Gelin B. R. & Karplus M. Dynamics of Folded Proteins. Nature 267(5612), 585–590 (1977). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
