

Intravenous immunglobulin binds beta amyloid and modifies its aggregation, neurotoxicity and microglial phagocytosis in vitro
- PMID: 23696796
- PMCID: PMC3656042
- DOI: 10.1371/journal.pone.0063162
Intravenous immunglobulin binds beta amyloid and modifies its aggregation, neurotoxicity and microglial phagocytosis in vitro
Abstract
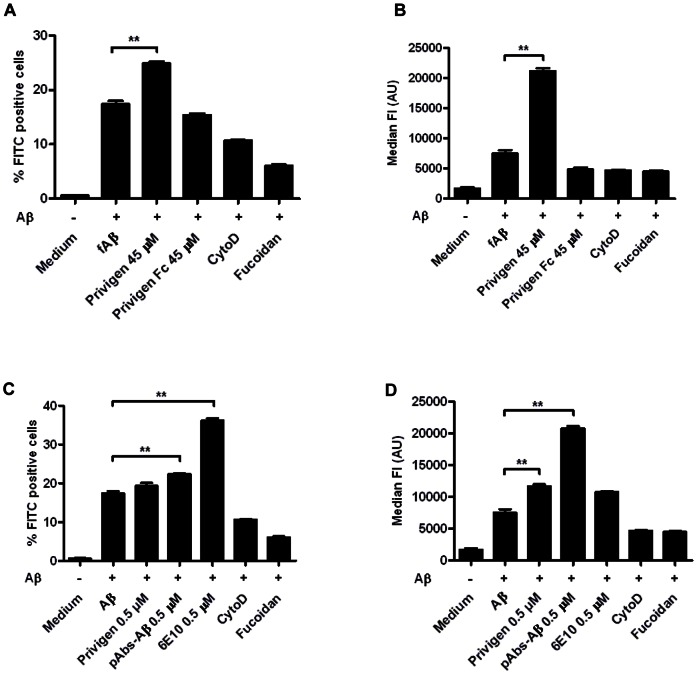
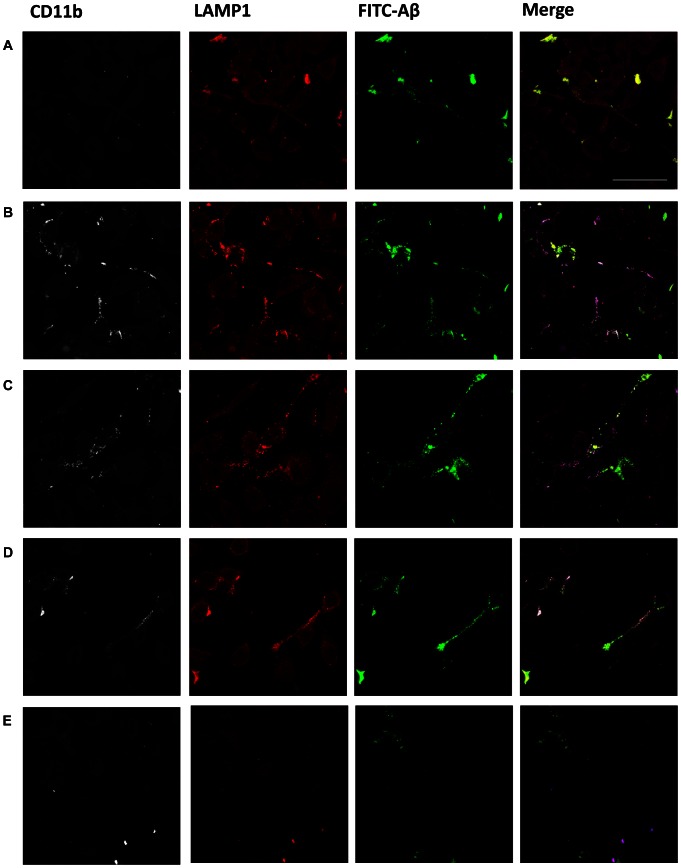
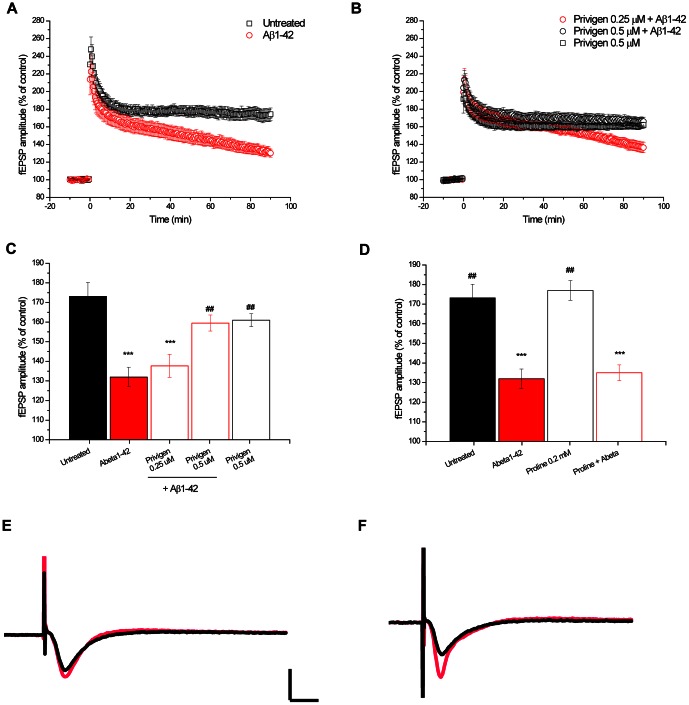
Intravenous Immunoglobulin (IVIG) has been proposed as a potential therapeutic for Alzheimer's disease (AD) and its efficacy is currently being tested in mild-to-moderate AD. Earlier studies reported the presence of anti-amyloid beta (Aβ) antibodies in IVIG. These observations led to clinical studies investigating the potential role of IVIG as a therapeutic agent in AD. Also, IVIG is known to mediate beneficial effects in chronic inflammatory and autoimmune conditions by interfering with various pathological processes. Therefore, we investigated the effects of IVIG and purified polyclonal Aβ-specific antibodies (pAbs-Aβ) on aggregation, toxicity and phagocytosis of Aβ in vitro, thus elucidating some of the potential mechanisms of action of IVIG in AD patients. We report that both IVIG and pAbs-Aβ specifically bound to Aβ and inhibited its aggregation in a dose-dependent manner as measured by Thioflavin T assay. Additionally, IVIG and the purified pAbs-Aβ inhibited Aβ-induced neurotoxicity in the SH-SY5Y human neuroblastoma cell line and prevented Aβ binding to rat primary cortical neurons. Interestingly, IVIG and pAbs-Aβ also increased the number of phagocytosing cells as well as the amount of phagocytosed fibrillar Aβ by BV-2 microglia. Phagocytosis of Aβ depended on receptor-mediated endocytosis and was accompanied by upregulation of CD11b expression. Importantly, we could also show that Privigen dose-dependently reversed Aβ-mediated LTP inhibition in mouse hippocampal slices. Therefore, our in vitro results suggest that IVIG may have an impact on different processes involved in AD pathogenesis, thereby promoting further understanding of the effects of IVIG observed in clinical studies.
Conflict of interest statement
Figures
Similar articles
-
Human intravenous immunoglobulin provides protection against Aβ toxicity by multiple mechanisms in a mouse model of Alzheimer's disease.J Neuroinflammation. 2010 Dec 7;7:90. doi: 10.1186/1742-2094-7-90. J Neuroinflammation. 2010. PMID: 21138577 Free PMC article.
-
Fibrillar Aβ triggers microglial proteome alterations and dysfunction in Alzheimer mouse models.Elife. 2020 Jun 8;9:e54083. doi: 10.7554/eLife.54083. Elife. 2020. PMID: 32510331 Free PMC article.
-
Poly(ADP-ribose)polymerase-1 modulates microglial responses to amyloid β.J Neuroinflammation. 2011 Nov 3;8:152. doi: 10.1186/1742-2094-8-152. J Neuroinflammation. 2011. PMID: 22051244 Free PMC article.
-
Intravenous immunoglobulins for Alzheimer's disease.Curr Alzheimer Res. 2014;11(7):626-36. doi: 10.2174/1567205011666140812113415. Curr Alzheimer Res. 2014. PMID: 25115546 Review.
-
Intravenous immunoglobulin and Alzheimer's disease immunotherapy.Curr Opin Mol Ther. 2007 Feb;9(1):79-85. Curr Opin Mol Ther. 2007. PMID: 17330405 Review.
Cited by
-
Effects of ganoderic acid A on lipopolysaccharide-induced proinflammatory cytokine release from primary mouse microglia cultures.Exp Ther Med. 2018 Jan;15(1):847-853. doi: 10.3892/etm.2017.5472. Epub 2017 Nov 8. Exp Ther Med. 2018. PMID: 29399089 Free PMC article.
-
Extending the functional characteristics of naturally occurring autoantibodies against β-Amyloid, Prion Protein and α-Synuclein.PLoS One. 2018 Aug 29;13(8):e0202954. doi: 10.1371/journal.pone.0202954. eCollection 2018. PLoS One. 2018. PMID: 30157279 Free PMC article.
-
Effect of IVIG Formulation on IgG Binding to Self- and Exo- Antigens In Vitro and In Vivo.PLoS One. 2016 Aug 25;11(8):e0161826. doi: 10.1371/journal.pone.0161826. eCollection 2016. PLoS One. 2016. PMID: 27561008 Free PMC article.
References
-
- Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297: 353–356 297/5580/353 [pii]. doi:10.1126/science.1072994. - DOI - PubMed
-
- Weiner HL, Frenkel D (2006) Immunology and immunotherapy of Alzheimer's disease. Nat Rev Immunol 6: 404–416 nri1843 [pii]; doi:10.1038/nri1843. - DOI - PubMed
-
- Bard F, Cannon C, Barbour R, Burke RL, Games D, et al. (2000) Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med 6: 916–919 doi:10.1038/78682. - DOI - PubMed
-
- DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, et al. (2001) Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A 98: 8850–8855 151261398 [pii]. doi:10.1073/pnas.151261398. - DOI - PMC - PubMed
-
- Morgan D (2011) Immunotherapy for Alzheimer's disease. J Intern Med 269: 54–63 doi:10.1111/j.1365-2796.2010.02315.x. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
