

Rac-1 superactivation triggers insulin-independent glucose transporter 4 (GLUT4) translocation that bypasses signaling defects exerted by c-Jun N-terminal kinase (JNK)- and ceramide-induced insulin resistance
- PMID: 23640896
- PMCID: PMC3682551
- DOI: 10.1074/jbc.M113.467647
Rac-1 superactivation triggers insulin-independent glucose transporter 4 (GLUT4) translocation that bypasses signaling defects exerted by c-Jun N-terminal kinase (JNK)- and ceramide-induced insulin resistance
Abstract
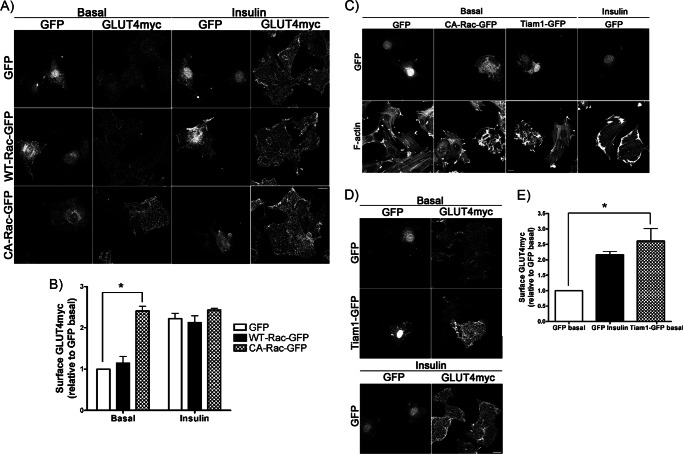
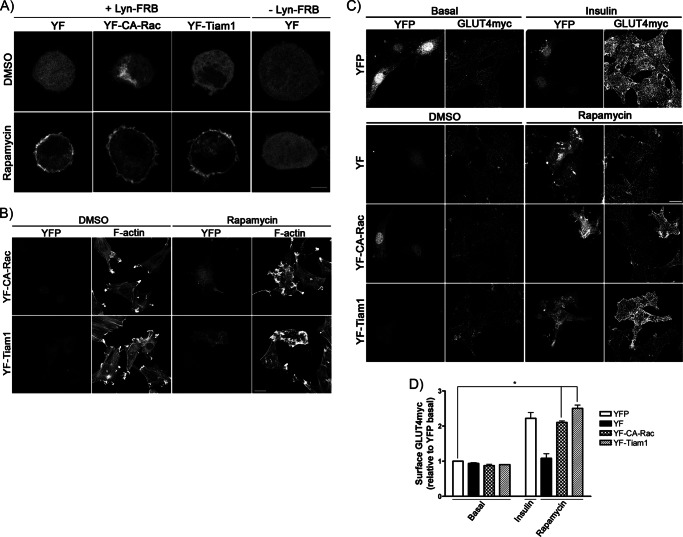
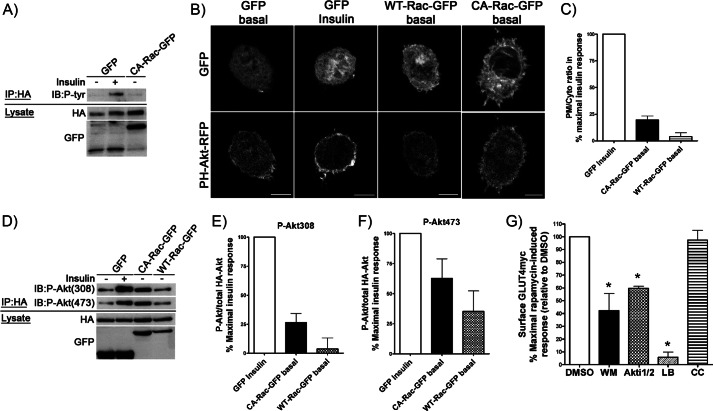
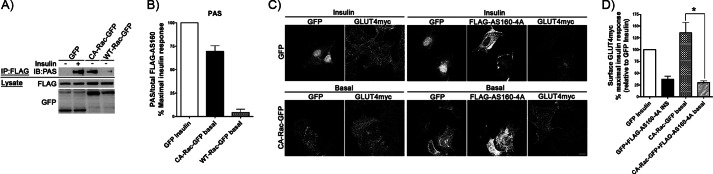
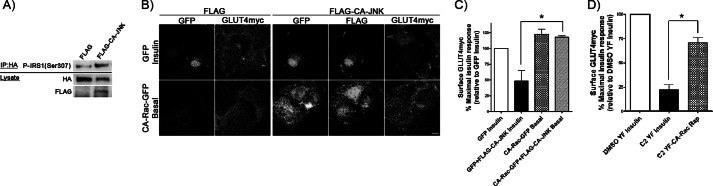
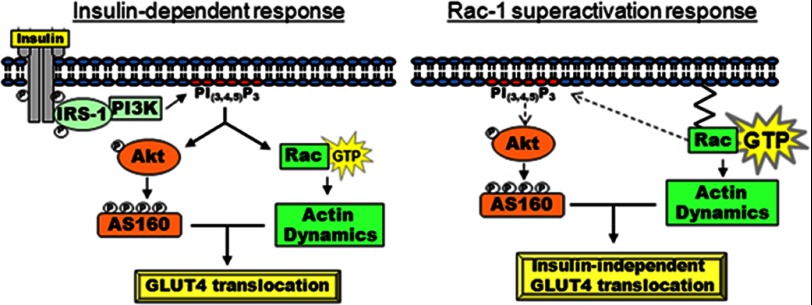
Insulin activates a cascade of signaling molecules, including Rac-1, Akt, and AS160, to promote the net gain of glucose transporter 4 (GLUT4) at the plasma membrane of muscle cells. Interestingly, constitutively active Rac-1 expression results in a hormone-independent increase in surface GLUT4; however, the molecular mechanism and significance behind this effect remain unresolved. Using L6 myoblasts stably expressing myc-tagged GLUT4, we found that overexpression of constitutively active but not wild-type Rac-1 sufficed to drive GLUT4 translocation to the membrane of comparable magnitude with that elicited by insulin. Stimulation of endogenous Rac-1 by Tiam1 overexpression elicited a similar hormone-independent gain in surface GLUT4. This effect on GLUT4 traffic could also be reproduced by acutely activating a Rac-1 construct via rapamycin-mediated heterodimerization. Strategies triggering Rac-1 "superactivation" (i.e. to levels above those attained by insulin alone) produced a modest gain in plasma membrane phosphatidylinositol 3,4,5-trisphosphate, moderate Akt activation, and substantial AS160 phosphorylation, which translated into GLUT4 translocation and negated the requirement for IRS-1. This unique signaling capacity exerted by Rac-1 superactivation bypassed the defects imposed by JNK- and ceramide-induced insulin resistance and allowed full and partial restoration of the GLUT4 translocation response, respectively. We propose that potent elevation of Rac-1 activation alone suffices to drive insulin-independent GLUT4 translocation in muscle cells, and such a strategy might be exploited to bypass signaling defects during insulin resistance.
Keywords: AS160; Akt; Ceramide; Glut4; Insulin Resistance; Jun N-terminal Kinase (JNK); Rac1.
Figures
Similar articles
-
Ceramide- and oxidant-induced insulin resistance involve loss of insulin-dependent Rac-activation and actin remodeling in muscle cells.Diabetes. 2007 Feb;56(2):394-403. doi: 10.2337/db06-0823. Diabetes. 2007. PMID: 17259384
-
Role of the guanine nucleotide exchange factor in Akt2-mediated plasma membrane translocation of GLUT4 in insulin-stimulated skeletal muscle.Cell Signal. 2014 Nov;26(11):2460-9. doi: 10.1016/j.cellsig.2014.07.002. Epub 2014 Jul 12. Cell Signal. 2014. PMID: 25025572
-
Activation of the small GTPase Rac1 by a specific guanine-nucleotide-exchange factor suffices to induce glucose uptake into skeletal-muscle cells.Biol Cell. 2008 Nov;100(11):645-57. doi: 10.1042/BC20070160. Biol Cell. 2008. PMID: 18482007
-
Small G proteins in insulin action: Rab and Rho families at the crossroads of signal transduction and GLUT4 vesicle traffic.Acta Physiol (Oxf). 2008 Jan;192(1):61-74. doi: 10.1111/j.1748-1716.2007.01778.x. Acta Physiol (Oxf). 2008. PMID: 18171430 Review.
-
Rac1 signalling towards GLUT4/glucose uptake in skeletal muscle.Cell Signal. 2011 Oct;23(10):1546-54. doi: 10.1016/j.cellsig.2011.05.022. Epub 2011 Jun 13. Cell Signal. 2011. PMID: 21683139 Review.
Cited by
-
Rho GTPases-Emerging Regulators of Glucose Homeostasis and Metabolic Health.Cells. 2019 May 9;8(5):434. doi: 10.3390/cells8050434. Cells. 2019. PMID: 31075957 Free PMC article. Review.
-
Certain Diet and Lifestyle May Contribute to Islet β-cells Protection in Type-2 Diabetes via the Modulation of Cellular PI3K/AKT Pathway.Open Biochem J. 2014 Nov 1;8:74-82. doi: 10.2174/1874091X01408010074. eCollection 2014. Open Biochem J. 2014. PMID: 25400709 Free PMC article.
-
Impaired Insulin Signaling Mediated by the Small GTPase Rac1 in Skeletal Muscle of the Leptin-Deficient Obese Mouse.Int J Mol Sci. 2023 Jul 16;24(14):11531. doi: 10.3390/ijms241411531. Int J Mol Sci. 2023. PMID: 37511290 Free PMC article.
-
A Boolean network of the crosstalk between IGF and Wnt signaling in aging satellite cells.PLoS One. 2018 Mar 29;13(3):e0195126. doi: 10.1371/journal.pone.0195126. eCollection 2018. PLoS One. 2018. PMID: 29596489 Free PMC article.
-
Rac1 Activation Caused by Membrane Translocation of a Guanine Nucleotide Exchange Factor in Akt2-Mediated Insulin Signaling in Mouse Skeletal Muscle.PLoS One. 2016 May 10;11(5):e0155292. doi: 10.1371/journal.pone.0155292. eCollection 2016. PLoS One. 2016. PMID: 27163697 Free PMC article.
References
-
- Baron A. D., Brechtel G., Wallace P., Edelman S. V. (1988) Rates and tissue sites of non-insulin- and insulin-mediated glucose uptake in humans. Am. J. Physiol. Endocrinol. Metab. 255, E769–E774 - PubMed
-
- Foley K., Boguslavsky S., Klip A. (2011) Endocytosis, recycling, and regulated exocytosis of glucose transporter 4. Biochemistry 50, 3048–3061 - PubMed
-
- Huang C., Thirone A. C., Huang X., Klip A. (2005) Differential contribution of insulin receptor substrates 1 versus 2 to insulin signaling and glucose uptake in l6 myotubes. J. Biol. Chem. 280, 19426–19435 - PubMed
-
- Somwar R., Niu W., Kim D. Y., Sweeney G., Randhawa V. K., Huang C., Ramlal T., Klip A. (2001) Differential effects of phosphatidylinositol 3-kinase inhibition on intracellular signals regulating GLUT4 translocation and glucose transport. J. Biol. Chem. 276, 46079–46087 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
