

Transposable elements are major contributors to the origin, diversification, and regulation of vertebrate long noncoding RNAs
- PMID: 23637635
- PMCID: PMC3636048
- DOI: 10.1371/journal.pgen.1003470
Transposable elements are major contributors to the origin, diversification, and regulation of vertebrate long noncoding RNAs
Abstract
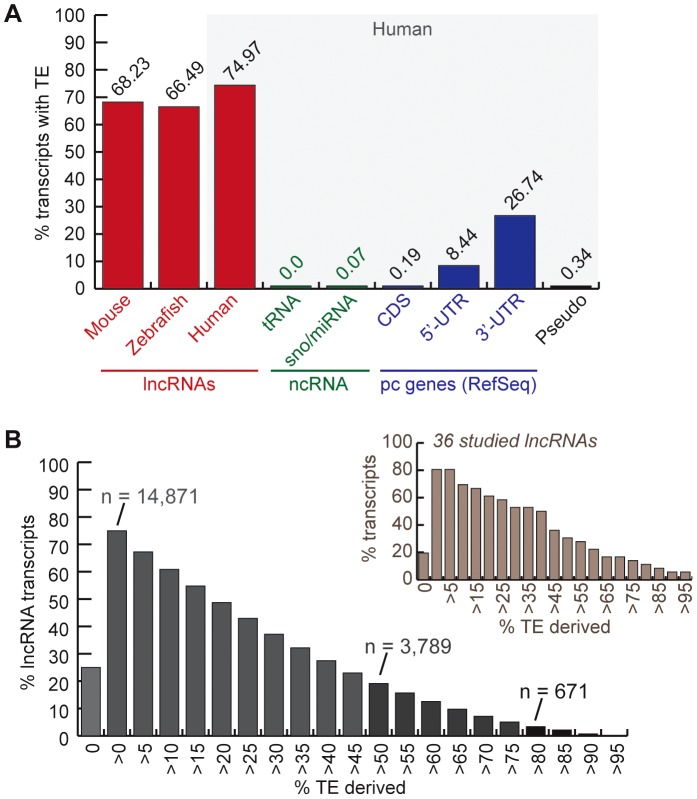
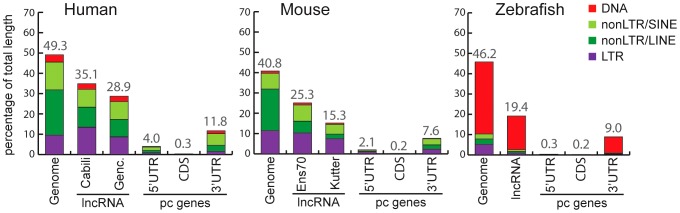
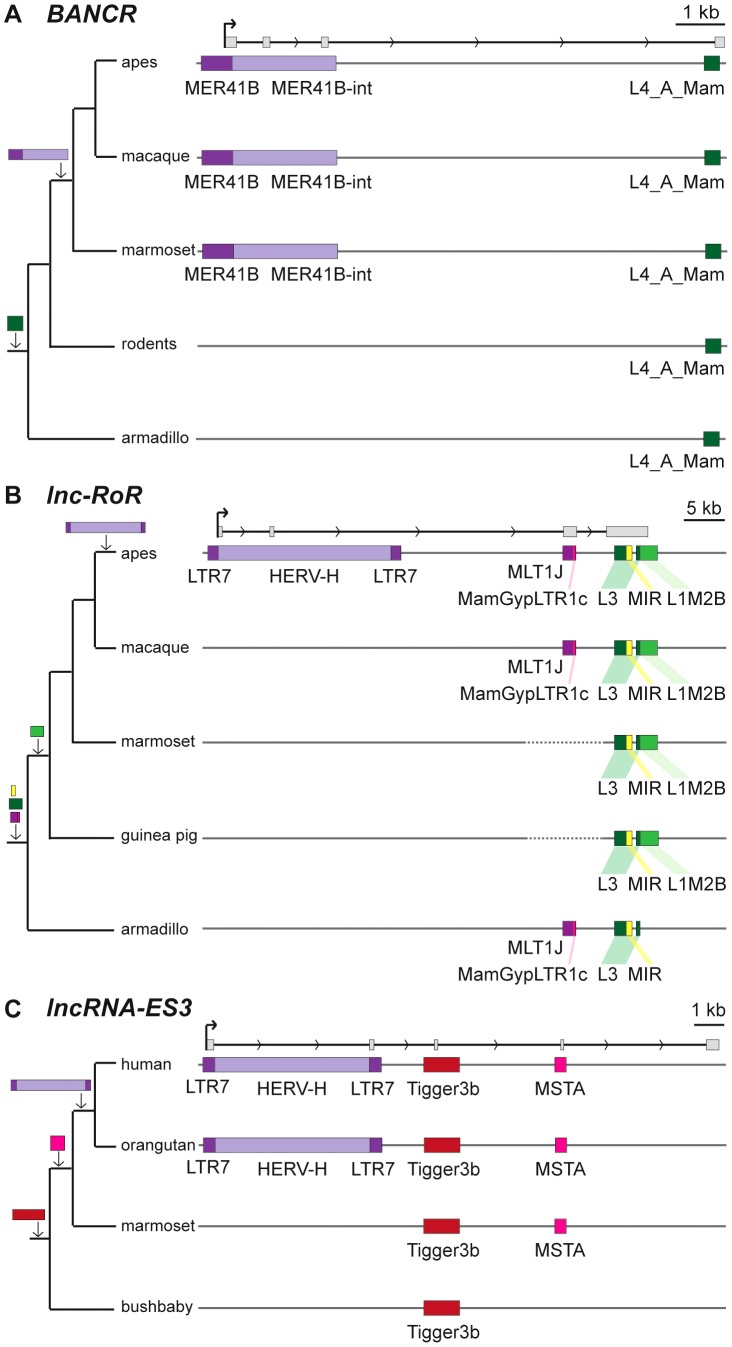
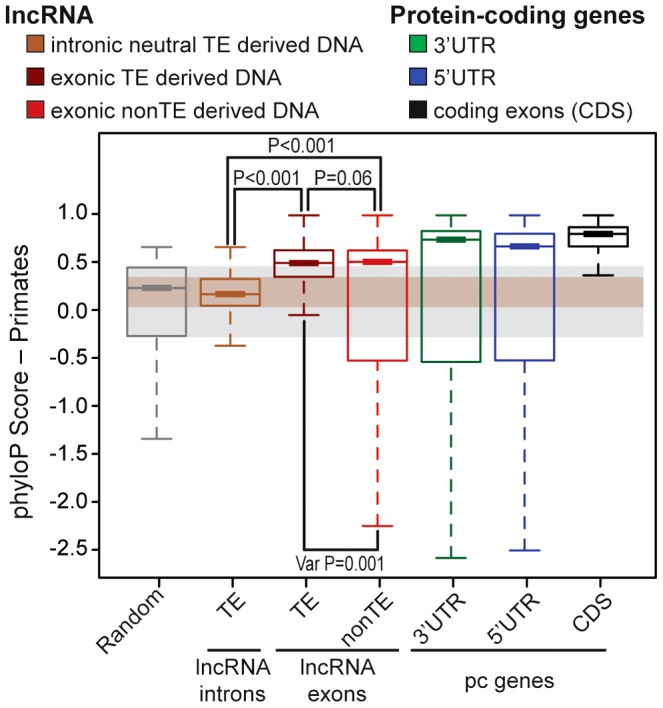
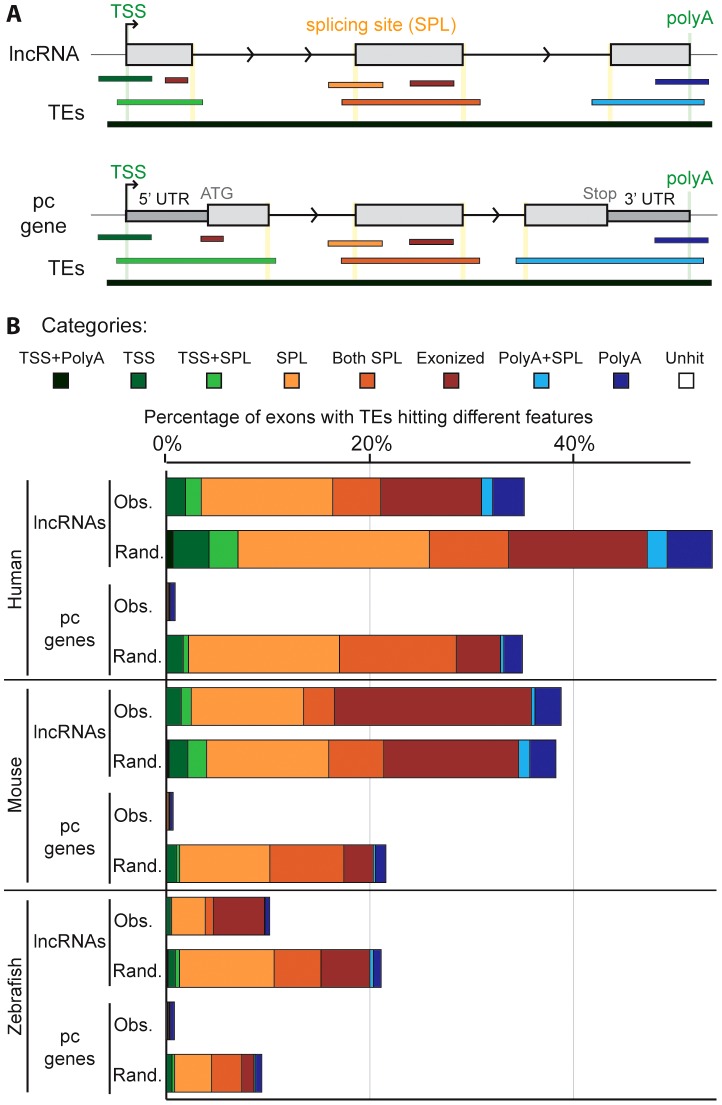
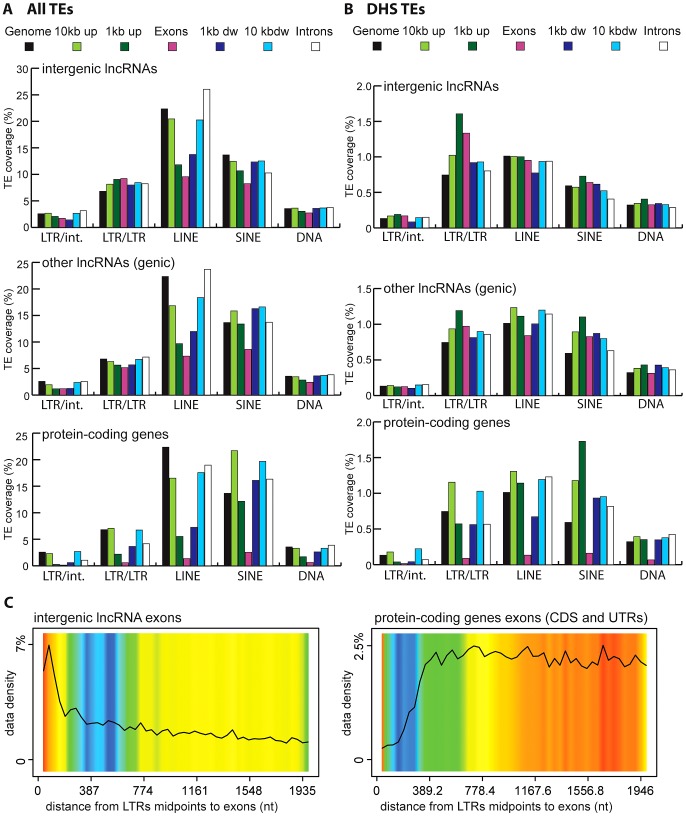
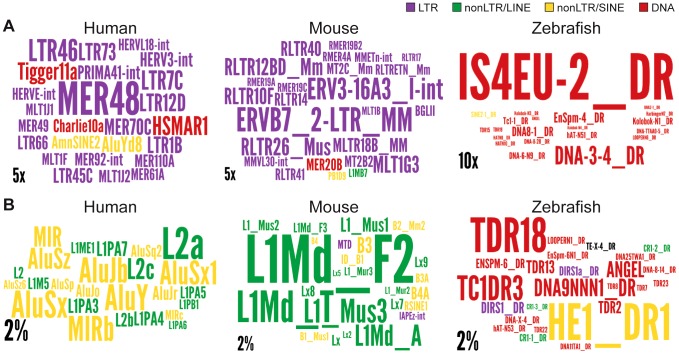
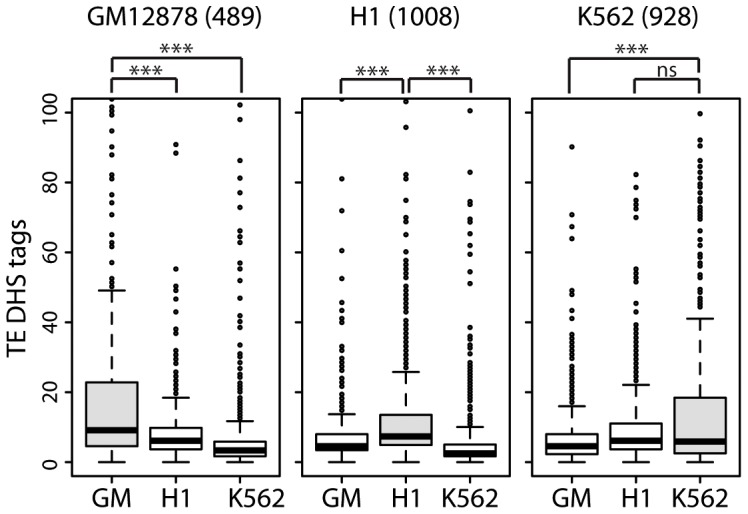
Advances in vertebrate genomics have uncovered thousands of loci encoding long noncoding RNAs (lncRNAs). While progress has been made in elucidating the regulatory functions of lncRNAs, little is known about their origins and evolution. Here we explore the contribution of transposable elements (TEs) to the makeup and regulation of lncRNAs in human, mouse, and zebrafish. Surprisingly, TEs occur in more than two thirds of mature lncRNA transcripts and account for a substantial portion of total lncRNA sequence (~30% in human), whereas they seldom occur in protein-coding transcripts. While TEs contribute less to lncRNA exons than expected, several TE families are strongly enriched in lncRNAs. There is also substantial interspecific variation in the coverage and types of TEs embedded in lncRNAs, partially reflecting differences in the TE landscapes of the genomes surveyed. In human, TE sequences in lncRNAs evolve under greater evolutionary constraint than their non-TE sequences, than their intronic TEs, or than random DNA. Consistent with functional constraint, we found that TEs contribute signals essential for the biogenesis of many lncRNAs, including ~30,000 unique sites for transcription initiation, splicing, or polyadenylation in human. In addition, we identified ~35,000 TEs marked as open chromatin located within 10 kb upstream of lncRNA genes. The density of these marks in one cell type correlate with elevated expression of the downstream lncRNA in the same cell type, suggesting that these TEs contribute to cis-regulation. These global trends are recapitulated in several lncRNAs with established functions. Finally a subset of TEs embedded in lncRNAs are subject to RNA editing and predicted to form secondary structures likely important for function. In conclusion, TEs are nearly ubiquitous in lncRNAs and have played an important role in the lineage-specific diversification of vertebrate lncRNA repertoires.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Expression and diversification analysis reveals transposable elements play important roles in the origin of Lycopersicon-specific lncRNAs in tomato.New Phytol. 2016 Mar;209(4):1442-55. doi: 10.1111/nph.13718. Epub 2015 Oct 23. New Phytol. 2016. PMID: 26494192
-
Mosquito long non-coding RNAs are enriched with Transposable Elements.Genet Mol Biol. 2022 Jan 24;45(1):e20210215. doi: 10.1590/1678-4685-GMB-2021-0215. eCollection 2022. Genet Mol Biol. 2022. PMID: 35088819 Free PMC article.
-
The RIDL hypothesis: transposable elements as functional domains of long noncoding RNAs.RNA. 2014 Jul;20(7):959-76. doi: 10.1261/rna.044560.114. Epub 2014 May 21. RNA. 2014. PMID: 24850885 Free PMC article.
-
The intertwining of transposable elements and non-coding RNAs.Int J Mol Sci. 2013 Jun 26;14(7):13307-28. doi: 10.3390/ijms140713307. Int J Mol Sci. 2013. PMID: 23803660 Free PMC article. Review.
-
Role of Transposable Elements in Gene Regulation in the Human Genome.Life (Basel). 2021 Feb 4;11(2):118. doi: 10.3390/life11020118. Life (Basel). 2021. PMID: 33557056 Free PMC article. Review.
Cited by
-
Activation of an endogenous retrovirus-associated long non-coding RNA in human adenocarcinoma.Genome Med. 2015 Mar 5;7(1):22. doi: 10.1186/s13073-015-0142-6. eCollection 2015. Genome Med. 2015. PMID: 25821520 Free PMC article.
-
Transposable Element Insertions in Long Intergenic Non-Coding RNA Genes.Front Bioeng Biotechnol. 2015 Jun 9;3:71. doi: 10.3389/fbioe.2015.00071. eCollection 2015. Front Bioeng Biotechnol. 2015. PMID: 26106594 Free PMC article.
-
The evolutionary landscape and expression pattern of plant lincRNAs.RNA Biol. 2022 Jan;19(1):1190-1207. doi: 10.1080/15476286.2022.2144609. RNA Biol. 2022. PMID: 36382947 Free PMC article.
-
Novel function of U7 snRNA in the repression of HERV1/LTR12s and lincRNAs in human cells.Nucleic Acids Res. 2024 Sep 23;52(17):10504-10519. doi: 10.1093/nar/gkae738. Nucleic Acids Res. 2024. PMID: 39189459 Free PMC article.
-
Exploration on cold adaptation of Antarctic lichen via detection of positive selection genes.IMA Fungus. 2024 Sep 9;15(1):29. doi: 10.1186/s43008-024-00160-x. IMA Fungus. 2024. PMID: 39252145 Free PMC article.
References
-
- Pheasant M, Mattick JS (2007) Raising the estimate of functional human sequences. Genome Res 17: 1245–1253. - PubMed
-
- Dinger ME, Amaral PP, Mercer TR, Mattick JS (2009) Pervasive transcription of the eukaryotic genome: functional indices and conceptual implications. Brief Funct Genomic Proteomic 8: 407–423. - PubMed
-
- Mercer TR, Dinger ME, Mattick JS (2009) Long non-coding RNAs: insights into functions. Nat Rev Genet 10: 155–159. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
