

p90 RSK2 mediates antianoikis signals by both transcription-dependent and -independent mechanisms
- PMID: 23608533
- PMCID: PMC3700113
- DOI: 10.1128/MCB.01677-12
p90 RSK2 mediates antianoikis signals by both transcription-dependent and -independent mechanisms
Abstract
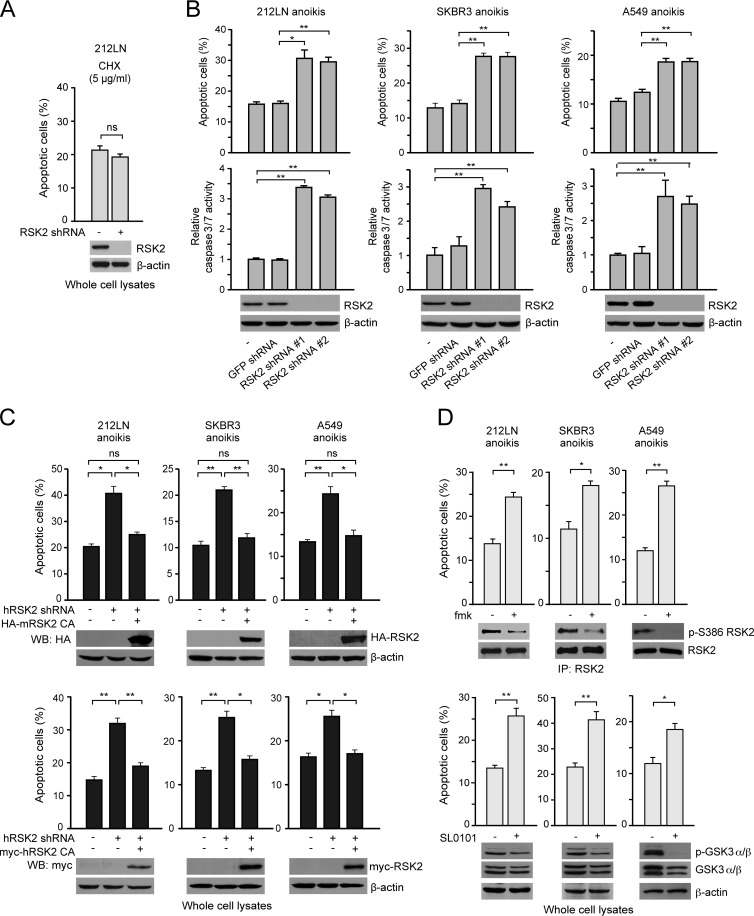
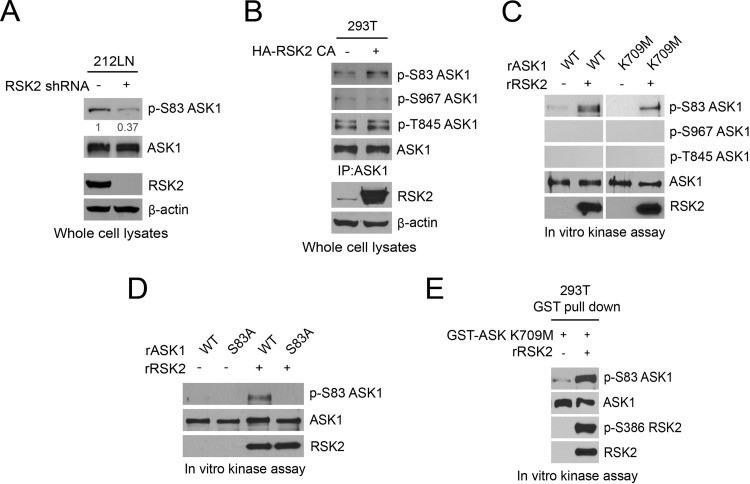
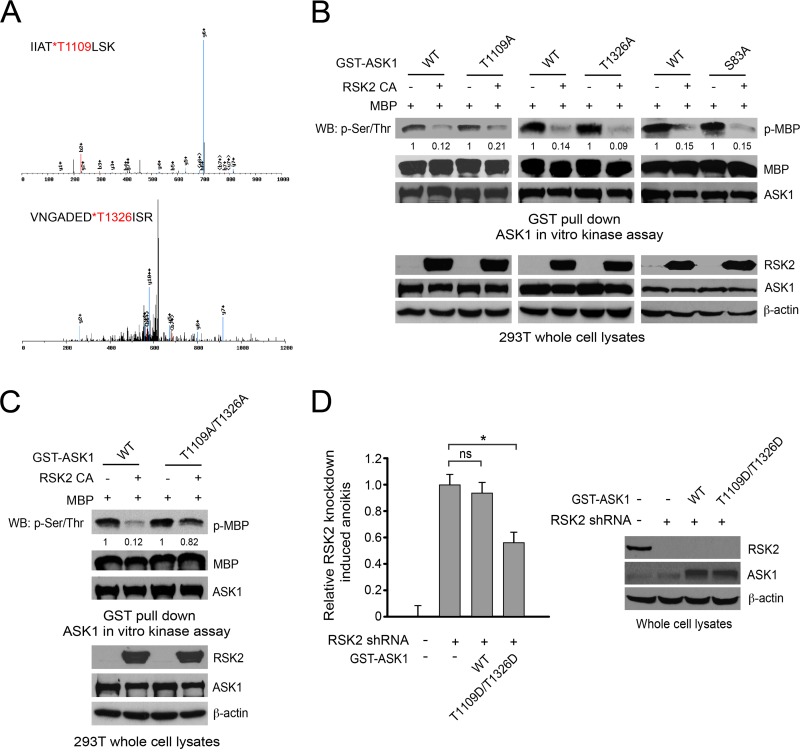
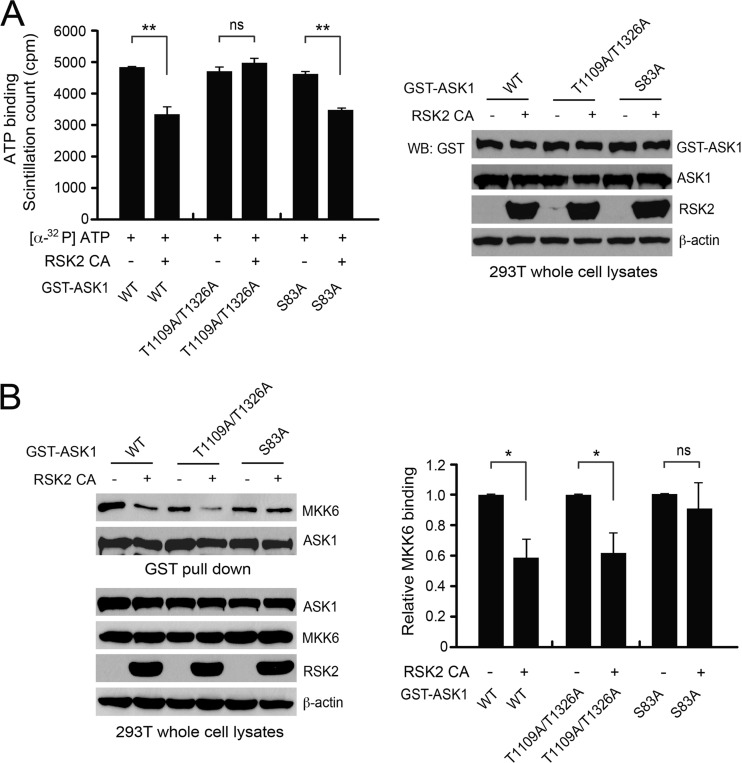
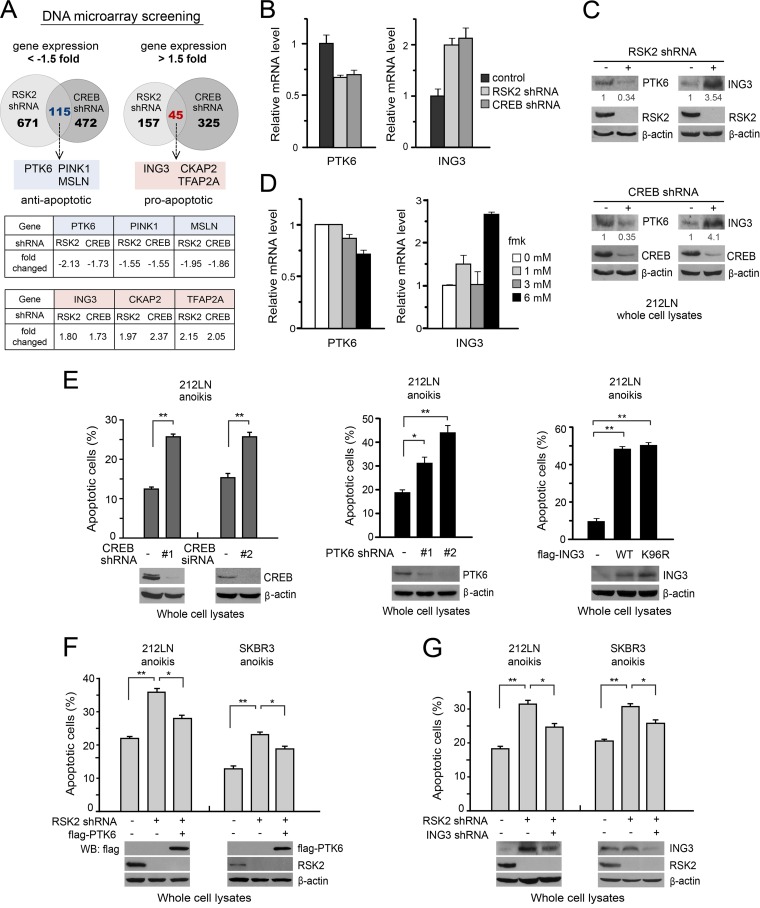
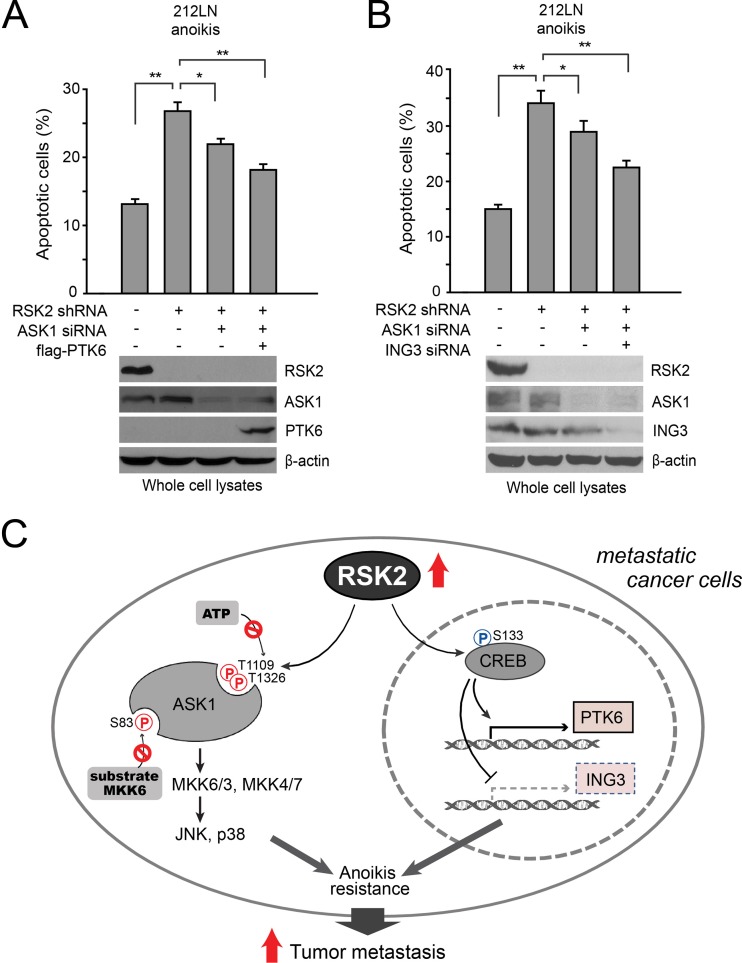
How invasive and metastatic tumor cells evade anoikis induction remains unclear. We found that knockdown of RSK2 sensitizes diverse cancer cells to anoikis induction, which is mediated through phosphorylation targets including apoptosis signal-regulating kinase 1 (ASK1) and cyclic AMP (cAMP) response element-binding protein (CREB). We provide evidence to show that RSK2 inhibits ASK1 by phosphorylating S83, T1109, and T1326 through a novel mechanism in which phospho-T1109/T1326 inhibits ATP binding to ASK1, while phospho-S83 attenuates ASK1 substrate MKK6 binding. Moreover, the RSK2→CREB signaling pathway provides antianoikis protection by regulating gene expression of protein effectors that are involved in cell death regulation, including the antiapoptotic factor protein tyrosine kinase 6 (PTK6) and the proapoptotic factor inhibitor-of-growth protein 3 (ING3). PTK6 overexpression or ING3 knockdown in addition to ASK1 knockdown further rescued the increased sensitivity to anoikis induction in RSK2 knockdown cells. These data together suggest that RSK2 functions as a signal integrator to provide antianoikis protection to cancer cells in both transcription-independent and -dependent manners, in part by signaling through ASK1 and CREB, and contributes to cancer cell invasion and tumor metastasis.
Figures

Similar articles
-
EGFR-phosphorylated GDH1 harmonizes with RSK2 to drive CREB activation and tumor metastasis in EGFR-activated lung cancer.Cell Rep. 2022 Dec 13;41(11):111827. doi: 10.1016/j.celrep.2022.111827. Cell Rep. 2022. PMID: 36516759 Free PMC article.
-
The prometastatic ribosomal S6 kinase 2-cAMP response element-binding protein (RSK2-CREB) signaling pathway up-regulates the actin-binding protein fascin-1 to promote tumor metastasis.J Biol Chem. 2013 Nov 8;288(45):32528-32538. doi: 10.1074/jbc.M113.500561. Epub 2013 Oct 1. J Biol Chem. 2013. PMID: 24085294 Free PMC article.
-
Epidermal growth factor stimulates RSK2 activation through activation of the MEK/ERK pathway and src-dependent tyrosine phosphorylation of RSK2 at Tyr-529.J Biol Chem. 2008 Feb 22;283(8):4652-7. doi: 10.1074/jbc.M709673200. Epub 2007 Dec 21. J Biol Chem. 2008. PMID: 18156174 Free PMC article.
-
RSK2 and its binding partners in cell proliferation, transformation and cancer development.Arch Pharm Res. 2017 Mar;40(3):291-303. doi: 10.1007/s12272-016-0880-z. Epub 2016 Dec 24. Arch Pharm Res. 2017. PMID: 28013489 Review.
-
Molecular Targeting of ERKs/RSK2 Signaling in Cancers.Curr Pharm Des. 2017 Nov 16;23(29):4247-4258. doi: 10.2174/1381612823666170714142338. Curr Pharm Des. 2017. PMID: 28714417 Review.
Cited by
-
EGFR-phosphorylated GDH1 harmonizes with RSK2 to drive CREB activation and tumor metastasis in EGFR-activated lung cancer.Cell Rep. 2022 Dec 13;41(11):111827. doi: 10.1016/j.celrep.2022.111827. Cell Rep. 2022. PMID: 36516759 Free PMC article.
-
Crystallographic mining of ASK1 regulators to unravel the intricate PPI interfaces for the discovery of small molecule.Comput Struct Biotechnol J. 2022 Jul 11;20:3734-3754. doi: 10.1016/j.csbj.2022.07.008. eCollection 2022. Comput Struct Biotechnol J. 2022. PMID: 35891784 Free PMC article. Review.
-
A short peptide LINC00665_18aa encoded by lncRNA LINC00665 suppresses the proliferation and migration of osteosarcoma cells through the regulation of the CREB1/RPS6KA3 interaction.PLoS One. 2023 Jun 7;18(6):e0286422. doi: 10.1371/journal.pone.0286422. eCollection 2023. PLoS One. 2023. PMID: 37285335 Free PMC article.
-
Succinyl-CoA ligase ADP-forming subunit beta promotes stress granule assembly to regulate redox and drive cancer metastasis.Proc Natl Acad Sci U S A. 2023 Jun 6;120(23):e2217332120. doi: 10.1073/pnas.2217332120. Epub 2023 May 30. Proc Natl Acad Sci U S A. 2023. PMID: 37253003 Free PMC article.
-
RSK promotes prostate cancer progression in bone through ING3, CKAP2, and PTK6-mediated cell survival.Mol Cancer Res. 2015 Feb;13(2):348-57. doi: 10.1158/1541-7786.MCR-14-0384-T. Epub 2014 Sep 4. Mol Cancer Res. 2015. PMID: 25189355 Free PMC article.
References
-
- Gupta GP, Massague J. 2006. Cancer metastasis: building a framework. Cell 127:679–695 - PubMed
-
- Fidler IJ. 2003. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat. Rev. Cancer 3:453–458 - PubMed
-
- Kang S, Elf S, Lythgoe K, Hitosugi T, Taunton J, Zhou W, Xiong L, Wang D, Muller S, Fan S, Sun SY, Marcus AI, Gu TL, Polakiewicz RD, Chen GZ, Khuri FR, Shin DM, Chen J. 2010. p90 ribosomal S6 kinase 2 promotes invasion and metastasis of human head and neck squamous cell carcinoma cells. J. Clin. Invest. 120:1165–1177 - PMC - PubMed
-
- Frodin M, Gammeltoft S. 1999. Role and regulation of 90 kDa ribosomal S6 kinase (RSK) in signal transduction. Mol. Cell. Endocrinol. 151:65–77 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous