

mTORC1 inhibition induces pain via IRS-1-dependent feedback activation of ERK
- PMID: 23607966
- PMCID: PMC3742001
- DOI: 10.1016/j.pain.2013.03.021
mTORC1 inhibition induces pain via IRS-1-dependent feedback activation of ERK
Abstract
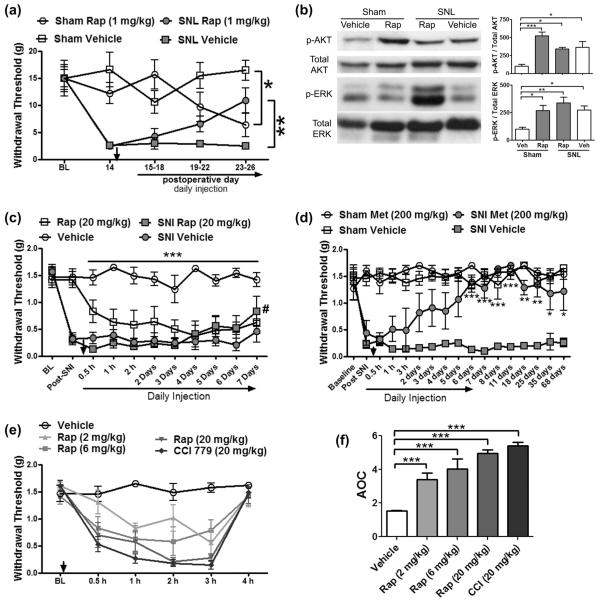
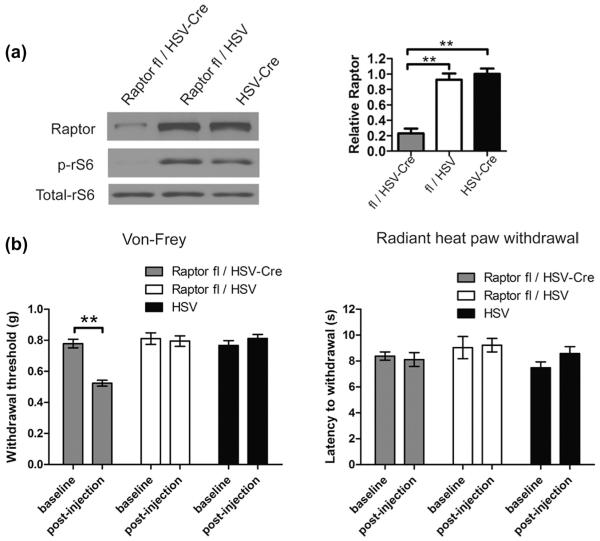
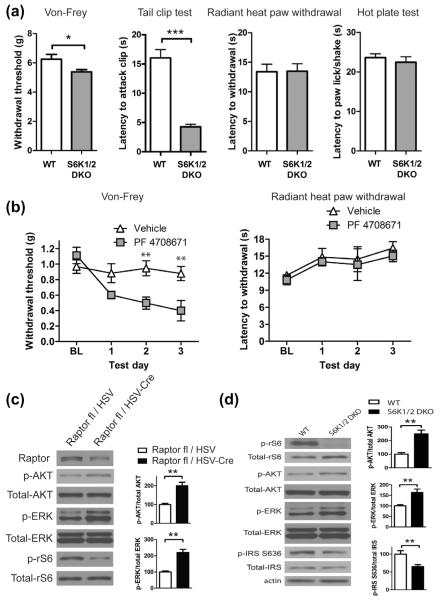
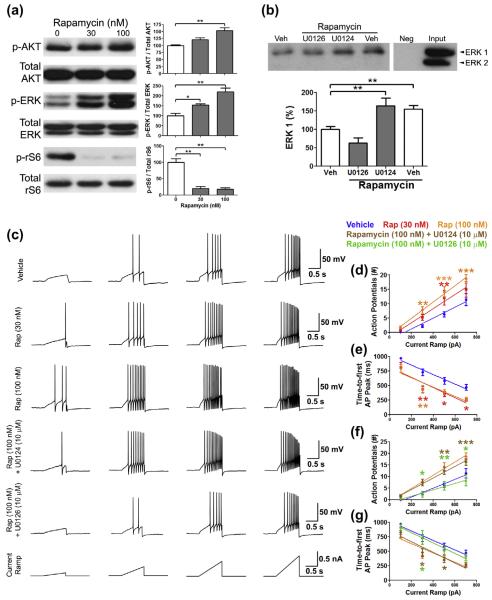
Mammalian target of rapamycin complex 1 (mTORC1) inhibitors are extensively used as immunosuppressants to prevent transplant rejection and in treatment of certain cancers. In patients, chronic treatment with rapamycin or its analogues (rapalogues) has been reported to lead to sensory hypersensitivity and pain conditions via an unknown mechanism. Here, we show that pharmacological or genetic inhibition of mTORC1 activates the extracellular signal-regulated kinase (ERK) pathway in sensory neurons via suppression of S6K1 to insulin receptor substrate 1 negative feedback loop. As a result, increased ERK activity induces sensory neuron sensitization, mechanical hypersensitivity, and spontaneous pain. The clinically available adenosine monophosphate-activated protein kinase activator, metformin, which is an antidiabetic drug, prevents rapamycin-induced ERK activation and the development of mechanical hypersensitivity and spontaneous pain. Taken together, our findings demonstrate that activation of the ERK pathway in sensory neurons as a consequence of mTORC1 inhibition leads to the development of pain. Importantly, this effect is abolished by co-treatment with metformin, thus providing a potential treatment option for rapalogue-evoked pain. Our findings highlight the physiological relevance of feedback signaling through mTORC1 inhibition and have important implications for development of pain therapeutics that target the mTOR pathway.
Copyright © 2013 International Association for the Study of Pain. Published by Elsevier B.V. All rights reserved.
Figures

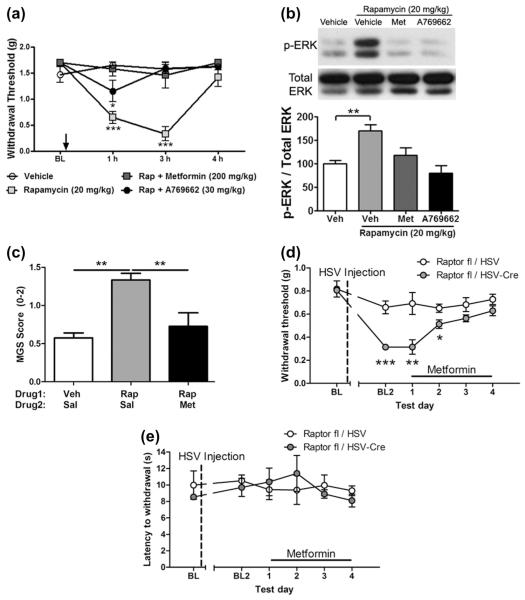
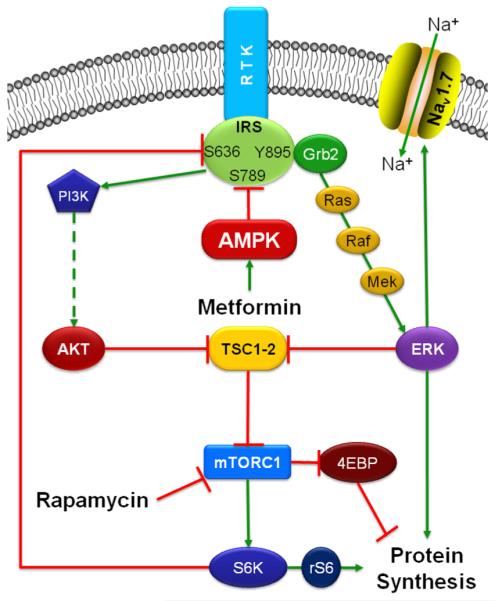
Similar articles
-
Chronic inhibition of the mTORC1/S6K1 pathway increases insulin-induced PI3K activity but inhibits Akt2 and glucose transport stimulation in 3T3-L1 adipocytes.Mol Endocrinol. 2010 Apr;24(4):766-78. doi: 10.1210/me.2009-0328. Epub 2010 Mar 4. Mol Endocrinol. 2010. PMID: 20203102 Free PMC article.
-
Alpha-synuclein overexpression negatively regulates insulin receptor substrate 1 by activating mTORC1/S6K1 signaling.Int J Biochem Cell Biol. 2015 Jul;64:25-33. doi: 10.1016/j.biocel.2015.03.006. Epub 2015 Mar 23. Int J Biochem Cell Biol. 2015. PMID: 25813876
-
mTOR complex 2 regulates proper turnover of insulin receptor substrate-1 via the ubiquitin ligase subunit Fbw8.Mol Cell. 2012 Dec 28;48(6):875-87. doi: 10.1016/j.molcel.2012.09.029. Epub 2012 Nov 8. Mol Cell. 2012. PMID: 23142081 Free PMC article.
-
Deconstructing feedback-signaling networks to improve anticancer therapy with mTORC1 inhibitors.Cell Cycle. 2008 Dec 15;7(24):3805-9. doi: 10.4161/cc.7.24.7244. Epub 2008 Dec 22. Cell Cycle. 2008. PMID: 19098454 Free PMC article. Review.
-
Regulation of insulin receptor substrate-1 by mTORC2 (mammalian target of rapamycin complex 2).Biochem Soc Trans. 2013 Aug;41(4):896-901. doi: 10.1042/BST20130018. Biochem Soc Trans. 2013. PMID: 23863152 Free PMC article. Review.
Cited by
-
The Antidiabetic Drug Metformin Regulates Voltage-Gated Sodium Channel NaV1.7 via the Ubiquitin-Ligase NEDD4-2.eNeuro. 2022 Mar 4;9(2):ENEURO.0409-21.2022. doi: 10.1523/ENEURO.0409-21.2022. Print 2022 Mar-Apr. eNeuro. 2022. PMID: 35131865 Free PMC article.
-
The use of metformin is associated with decreased lumbar radiculopathy pain.J Pain Res. 2013 Dec 9;6:755-63. doi: 10.2147/JPR.S52205. eCollection 2013. J Pain Res. 2013. PMID: 24357937 Free PMC article.
-
Fufang Kushen injection inhibits sarcoma growth and tumor-induced hyperalgesia via TRPV1 signaling pathways.Cancer Lett. 2014 Dec 28;355(2):232-41. doi: 10.1016/j.canlet.2014.08.037. Epub 2014 Sep 19. Cancer Lett. 2014. PMID: 25242356 Free PMC article.
-
mTOR-neuropeptide Y signaling sensitizes nociceptors to drive neuropathic pain.JCI Insight. 2022 Nov 22;7(22):e159247. doi: 10.1172/jci.insight.159247. JCI Insight. 2022. PMID: 36194480 Free PMC article.
-
Sex Differences in Neuropathy: The Paradigmatic Case of MetFormin.Int J Mol Sci. 2022 Nov 22;23(23):14503. doi: 10.3390/ijms232314503. Int J Mol Sci. 2022. PMID: 36498830 Free PMC article.
References
-
- Bailey CJ, Turner RC. Metformin. N Engl J Med. 1996;334:574–9. - PubMed
-
- Bourquin AF, Suveges M, Pertin M, Gilliard N, Sardy S, Davison AC, Spahn DR, Decosterd I. Assessment and analysis of mechanical allodynia-like behavior induced by spared nerve injury (SNI) in the mouse. PAIN®. 2006;122:e11–4. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
