

Inhibition of TFG function causes hereditary axon degeneration by impairing endoplasmic reticulum structure
- PMID: 23479643
- PMCID: PMC3612678
- DOI: 10.1073/pnas.1217197110
Inhibition of TFG function causes hereditary axon degeneration by impairing endoplasmic reticulum structure
Abstract
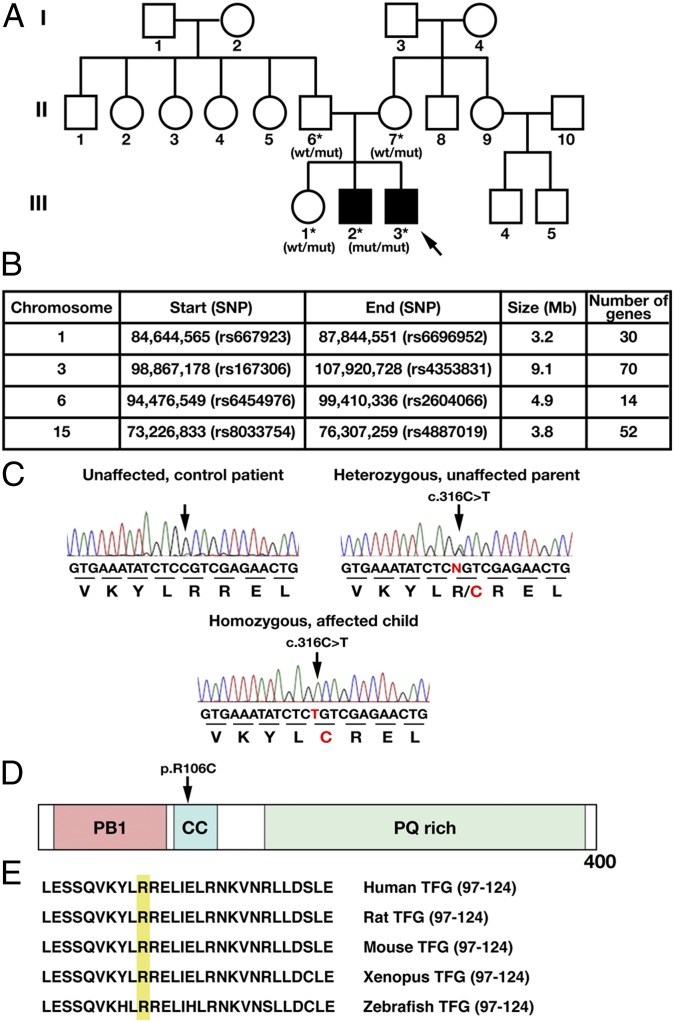
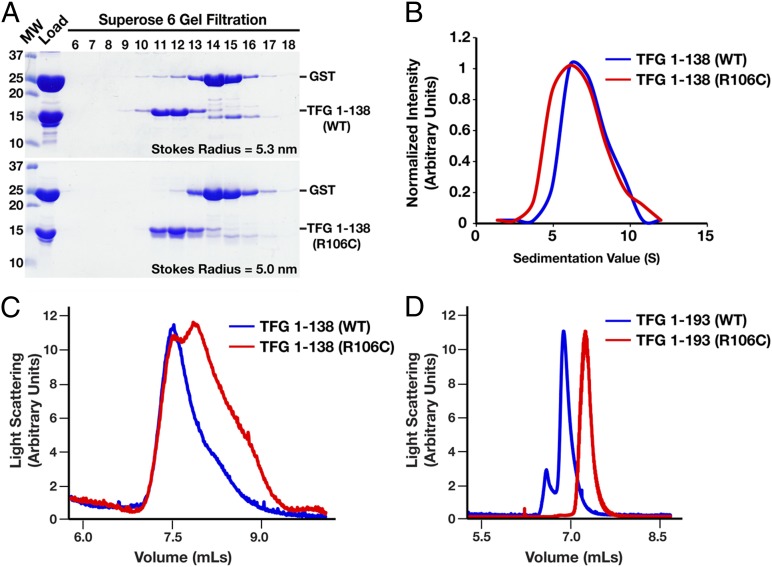
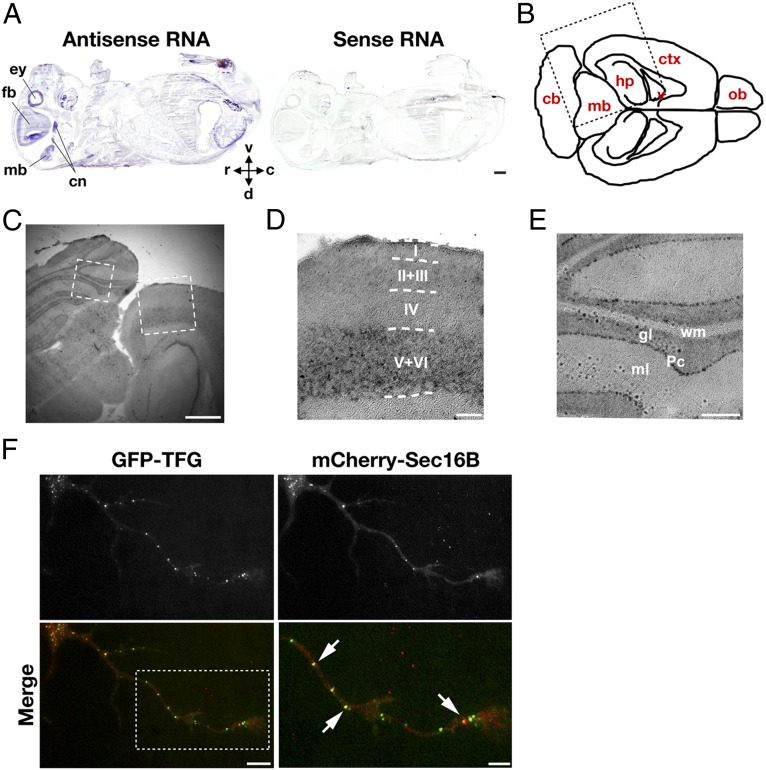
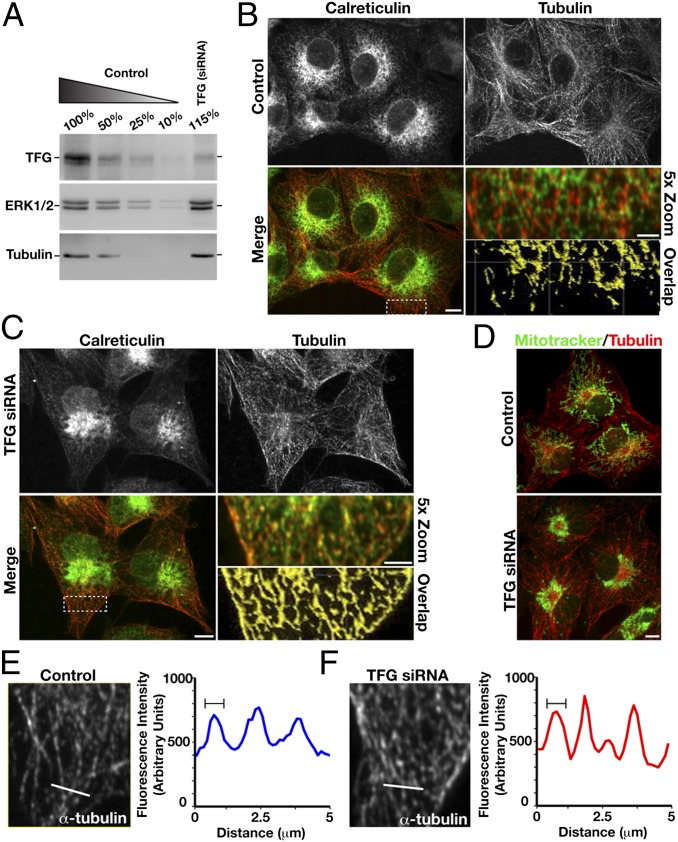
Hereditary spastic paraplegias are a clinically and genetically heterogeneous group of gait disorders. Their pathological hallmark is a length-dependent distal axonopathy of nerve fibers in the corticospinal tract. Involvement of other neurons can cause additional neurological symptoms, which define a diverse set of complex hereditary spastic paraplegias. We present two siblings who have the unusual combination of early-onset spastic paraplegia, optic atrophy, and neuropathy. Genome-wide SNP-typing, linkage analysis, and exome sequencing revealed a homozygous c.316C>T (p.R106C) variant in the Trk-fused gene (TFG) as the only plausible mutation. Biochemical characterization of the mutant protein demonstrated a defect in its ability to self-assemble into an oligomeric complex, which is critical for normal TFG function. In cell lines, TFG inhibition slows protein secretion from the endoplasmic reticulum (ER) and alters ER morphology, disrupting organization of peripheral ER tubules and causing collapse of the ER network onto the underlying microtubule cytoskeleton. The present study provides a unique link between altered ER architecture and neurodegeneration.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Pathogenic TFG Mutations Underlying Hereditary Spastic Paraplegia Impair Secretory Protein Trafficking and Axon Fasciculation.Cell Rep. 2018 Aug 28;24(9):2248-2260. doi: 10.1016/j.celrep.2018.07.081. Cell Rep. 2018. PMID: 30157421 Free PMC article.
-
Novel Genetic, Clinical, and Pathomechanistic Insights into TFG-Associated Hereditary Spastic Paraplegia.Hum Mutat. 2016 Nov;37(11):1157-1161. doi: 10.1002/humu.23060. Epub 2016 Aug 30. Hum Mutat. 2016. PMID: 27492651
-
Mutations in the ER-shaping protein reticulon 2 cause the axon-degenerative disorder hereditary spastic paraplegia type 12.J Clin Invest. 2012 Feb;122(2):538-44. doi: 10.1172/JCI60560. Epub 2012 Jan 9. J Clin Invest. 2012. PMID: 22232211 Free PMC article.
-
ER Morphology in the Pathogenesis of Hereditary Spastic Paraplegia.Cells. 2021 Oct 25;10(11):2870. doi: 10.3390/cells10112870. Cells. 2021. PMID: 34831093 Free PMC article. Review.
-
TFG-Related Neurologic Disorders: New Insights Into Relationships Between Endoplasmic Reticulum and Neurodegeneration.J Neuropathol Exp Neurol. 2016 Apr;75(4):299-305. doi: 10.1093/jnen/nlw009. Epub 2016 Mar 4. J Neuropathol Exp Neurol. 2016. PMID: 26945032 Review.
Cited by
-
Autophagy in major human diseases.EMBO J. 2021 Oct 1;40(19):e108863. doi: 10.15252/embj.2021108863. Epub 2021 Aug 30. EMBO J. 2021. PMID: 34459017 Free PMC article. Review.
-
STIM1 Is Required for Remodeling of the Endoplasmic Reticulum and Microtubule Cytoskeleton in Steering Growth Cones.J Neurosci. 2019 Jun 26;39(26):5095-5114. doi: 10.1523/JNEUROSCI.2496-18.2019. Epub 2019 Apr 25. J Neurosci. 2019. PMID: 31023836 Free PMC article.
-
Unraveling the Role of Heme in Neurodegeneration.Front Neurosci. 2018 Oct 9;12:712. doi: 10.3389/fnins.2018.00712. eCollection 2018. Front Neurosci. 2018. PMID: 30356807 Free PMC article. Review.
-
Involvement of neuronal and muscular Trk-fused gene (TFG) defects in the development of neurodegenerative diseases.Sci Rep. 2022 Feb 4;12(1):1966. doi: 10.1038/s41598-022-05884-7. Sci Rep. 2022. PMID: 35121777 Free PMC article.
-
Spatial and temporal immunoreactivity in the rat brain using an affinity purified polyclonal antibody to DNSP-11.J Chem Neuroanat. 2019 Oct;100:101664. doi: 10.1016/j.jchemneu.2019.101664. Epub 2019 Aug 5. J Chem Neuroanat. 2019. PMID: 31394198 Free PMC article.
References
-
- Schüle R, Schöls L. Genetics of hereditary spastic paraplegias. Semin Neurol. 2011;31(5):484–493. - PubMed
-
- Fink JK. Hereditary spastic paraplegia. Curr Neurol Neurosci Rep. 2006;6(1):65–76. - PubMed
-
- Salinas S, Proukakis C, Crosby A, Warner TT. Hereditary spastic paraplegia: clinical features and pathogenetic mechanisms. Lancet Neurol. 2008;7(12):1127–1138. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
