

Biophysical insights into how surfaces, including lipid membranes, modulate protein aggregation related to neurodegeneration
- PMID: 23459674
- PMCID: PMC3585431
- DOI: 10.3389/fneur.2013.00017
Biophysical insights into how surfaces, including lipid membranes, modulate protein aggregation related to neurodegeneration
Abstract
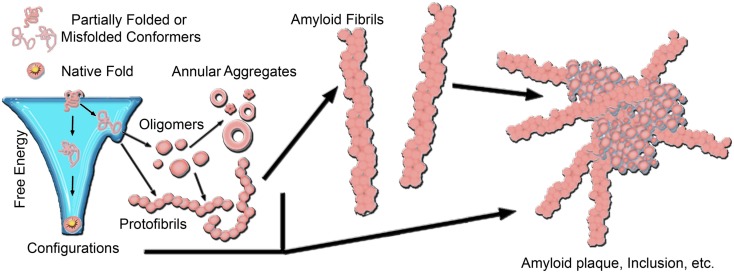
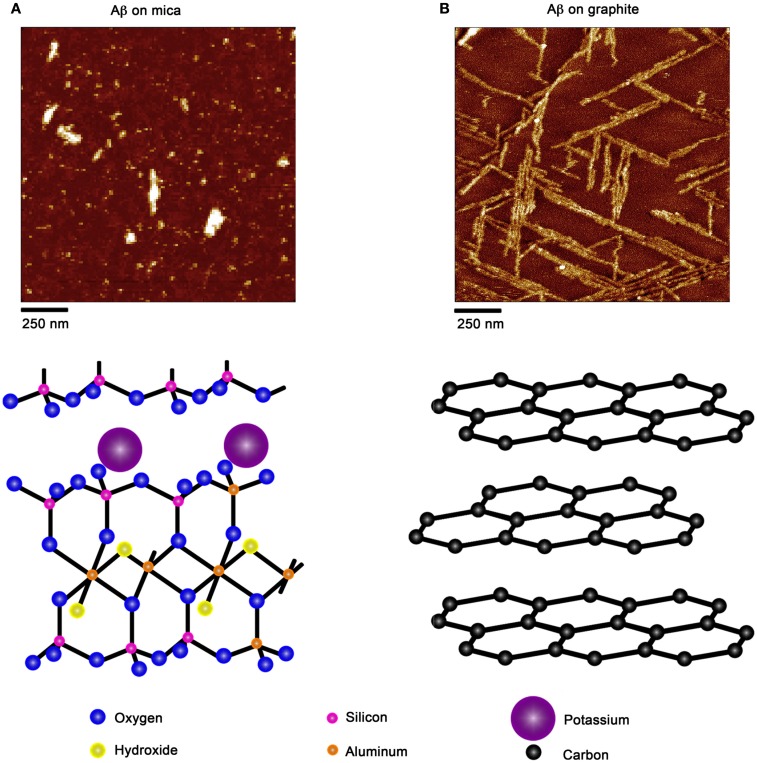
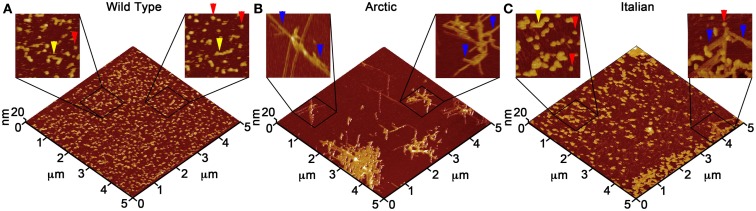
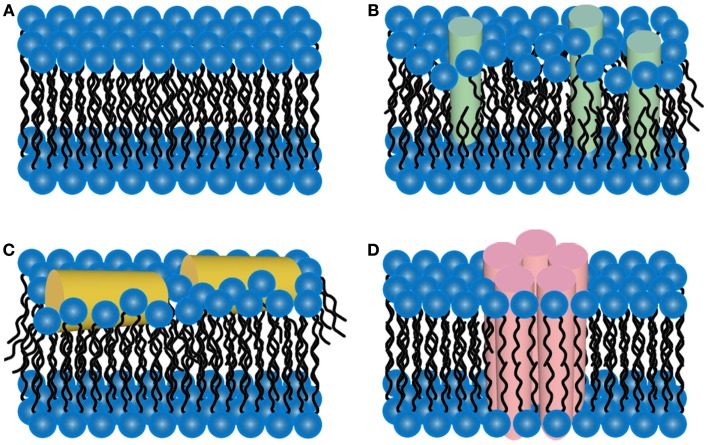
There are a vast number of neurodegenerative diseases, including Alzheimer's disease (AD), Parkinson's disease (PD), and Huntington's disease (HD), associated with the rearrangement of specific proteins to non-native conformations that promotes aggregation and deposition within tissues and/or cellular compartments. These diseases are commonly classified as protein-misfolding or amyloid diseases. The interaction of these proteins with liquid/surface interfaces is a fundamental phenomenon with potential implications for protein-misfolding diseases. Kinetic and thermodynamic studies indicate that significant conformational changes can be induced in proteins encountering surfaces, which can play a critical role in nucleating aggregate formation or stabilizing specific aggregation states. Surfaces of particular interest in neurodegenerative diseases are cellular and subcellular membranes that are predominately comprised of lipid components. The two-dimensional liquid environments provided by lipid bilayers can profoundly alter protein structure and dynamics by both specific and non-specific interactions. Importantly for misfolding diseases, these bilayer properties can not only modulate protein conformation, but also exert influence on aggregation state. A detailed understanding of the influence of (sub)cellular surfaces in driving protein aggregation and/or stabilizing specific aggregate forms could provide new insights into toxic mechanisms associated with these diseases. Here, we review the influence of surfaces in driving and stabilizing protein aggregation with a specific emphasis on lipid membranes.
Keywords: Alzheimer’s disease; Huntington’s disease; Parkinson’s disease; amyloid disease; lipid membranes; prion disease; protein aggregation.
Figures

Similar articles
-
Distinct role of hydration water in protein misfolding and aggregation revealed by fluctuating thermodynamics analysis.Acc Chem Res. 2015 Apr 21;48(4):956-65. doi: 10.1021/acs.accounts.5b00032. Epub 2015 Apr 6. Acc Chem Res. 2015. PMID: 25844814 Review.
-
Interaction of Amyloidogenic Proteins with Membranes and Molecular Mechanism for the Development of Alzheimer's disease.Alzheimers Res Ther Open Access. 2019;2(1):106. Epub 2019 Jun 6. Alzheimers Res Ther Open Access. 2019. PMID: 33135011 Free PMC article.
-
Lipid Membranes Influence the Ability of Small Molecules To Inhibit Huntingtin Fibrillization.Biochemistry. 2019 Oct 29;58(43):4361-4373. doi: 10.1021/acs.biochem.9b00739. Epub 2019 Oct 17. Biochemistry. 2019. PMID: 31608620 Free PMC article.
-
A review on protein misfolding, aggregation and strategies to prevent related ailments.Int J Biol Macromol. 2017 Dec;105(Pt 1):993-1000. doi: 10.1016/j.ijbiomac.2017.07.116. Epub 2017 Jul 23. Int J Biol Macromol. 2017. PMID: 28743576 Review.
-
Protein conformational misfolding and amyloid formation: characteristics of a new class of disorders that include Alzheimer's and Prion diseases.Curr Med Chem. 2002 Oct;9(19):1751-62. doi: 10.2174/0929867023369123. Curr Med Chem. 2002. PMID: 12369885 Review.
Cited by
-
Physical mechanisms of amyloid nucleation on fluid membranes.Proc Natl Acad Sci U S A. 2020 Dec 29;117(52):33090-33098. doi: 10.1073/pnas.2007694117. Epub 2020 Dec 16. Proc Natl Acad Sci U S A. 2020. PMID: 33328273 Free PMC article.
-
Deciphering the Proteotoxic Stress Responses Triggered by the Perturbed Thylakoid Proteostasis in Arabidopsis.Plants (Basel). 2021 Mar 10;10(3):519. doi: 10.3390/plants10030519. Plants (Basel). 2021. PMID: 33802194 Free PMC article.
-
Host-membrane interacting interface of the SARS coronavirus envelope protein: Immense functional potential of C-terminal domain.Biophys Chem. 2020 Nov;266:106452. doi: 10.1016/j.bpc.2020.106452. Epub 2020 Aug 11. Biophys Chem. 2020. PMID: 32818817 Free PMC article. Review.
-
Aggregation of an Amyloidogenic Peptide on Gold Surfaces.Biomolecules. 2023 Aug 18;13(8):1261. doi: 10.3390/biom13081261. Biomolecules. 2023. PMID: 37627326 Free PMC article.
-
Annexin B12 Trimer Formation is Governed by a Network of Protein-Protein and Protein-Lipid Interactions.Sci Rep. 2020 Mar 24;10(1):5301. doi: 10.1038/s41598-020-62343-x. Sci Rep. 2020. PMID: 32210350 Free PMC article.
References
LinkOut - more resources
Full Text Sources
Other Literature Sources