

Activation of PPARα ameliorates hepatic insulin resistance and steatosis in high fructose-fed mice despite increased endoplasmic reticulum stress
- PMID: 23349482
- PMCID: PMC3661626
- DOI: 10.2337/db12-1397
Activation of PPARα ameliorates hepatic insulin resistance and steatosis in high fructose-fed mice despite increased endoplasmic reticulum stress
Abstract
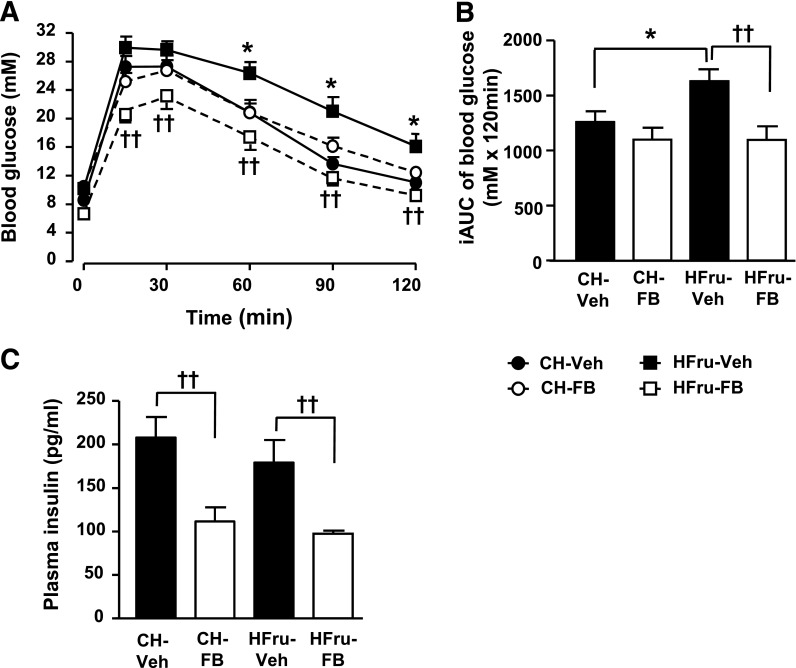
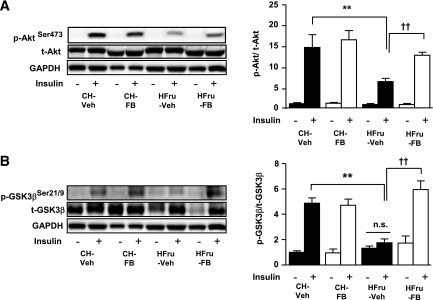
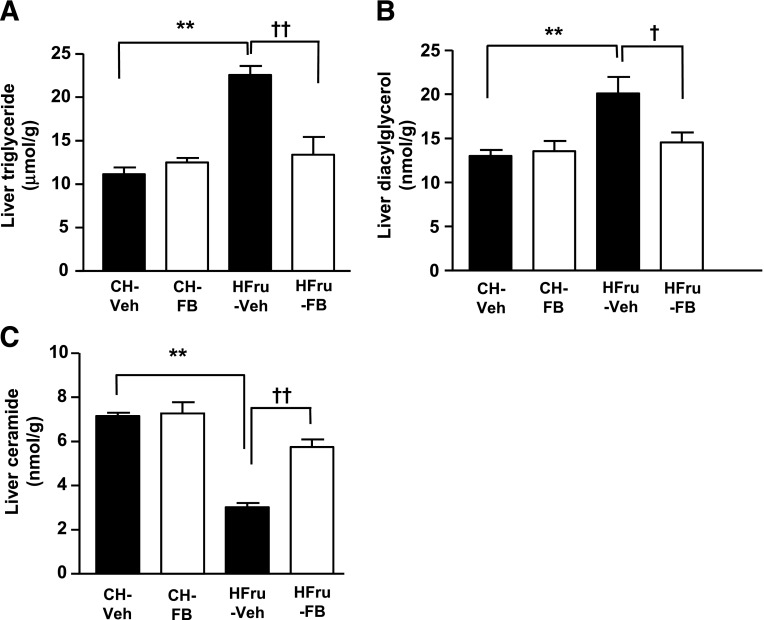
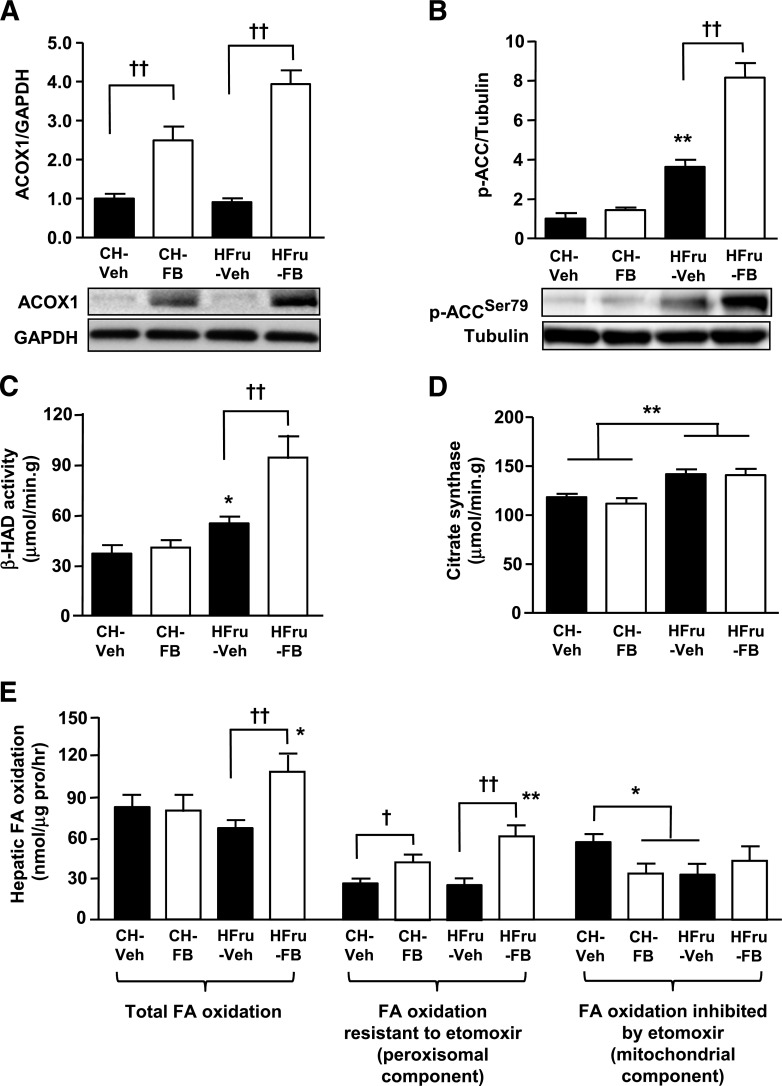
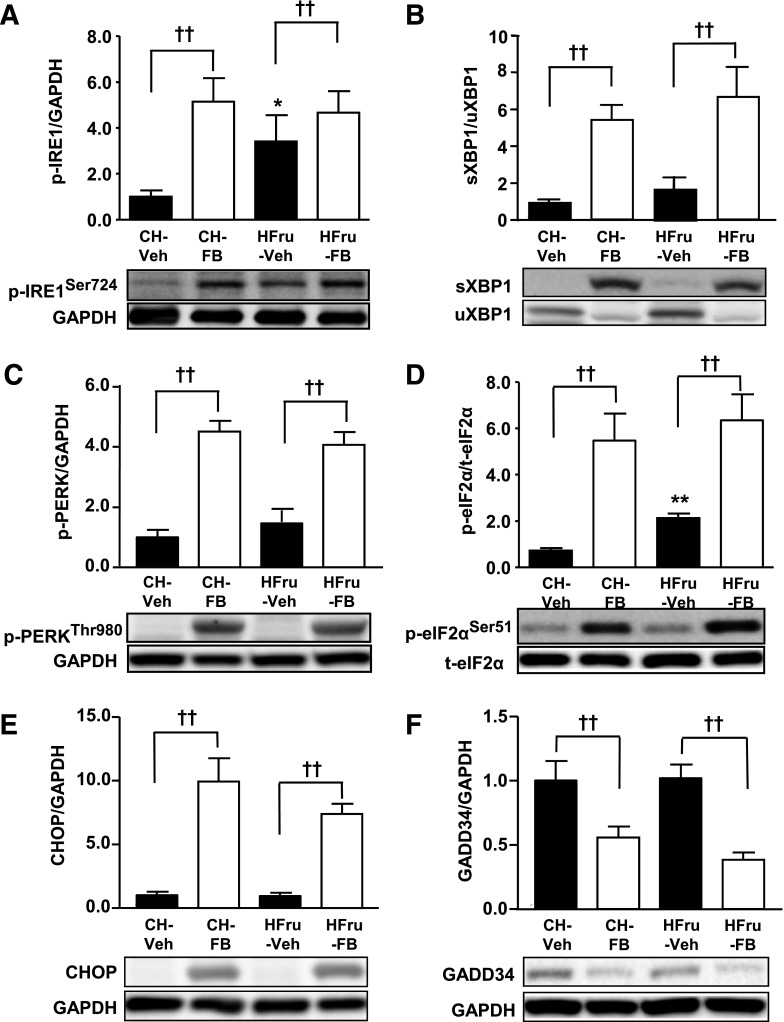
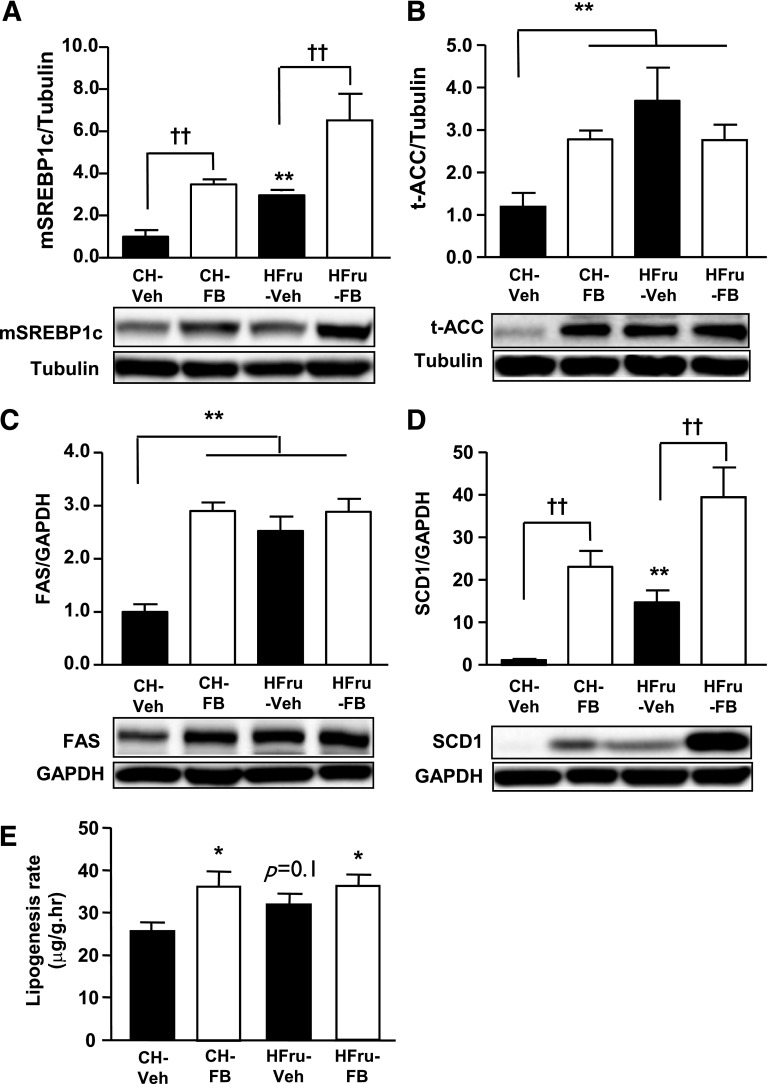
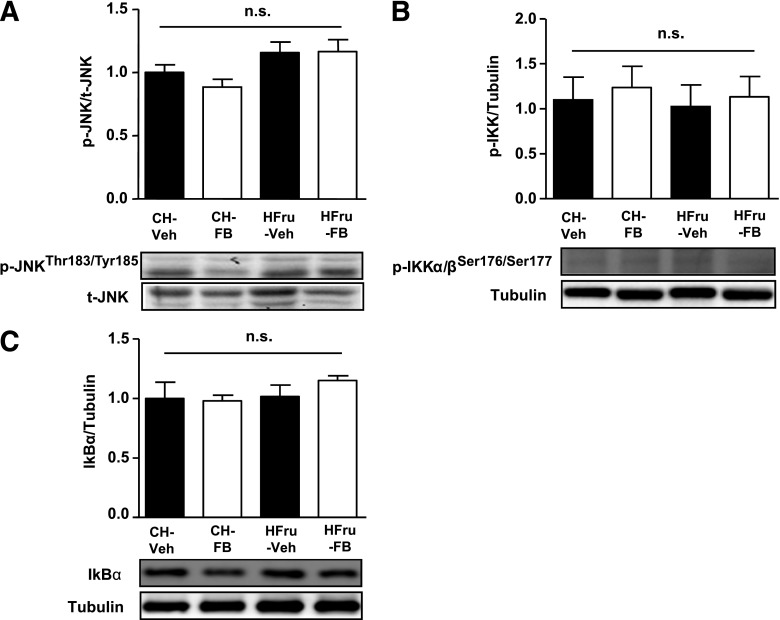
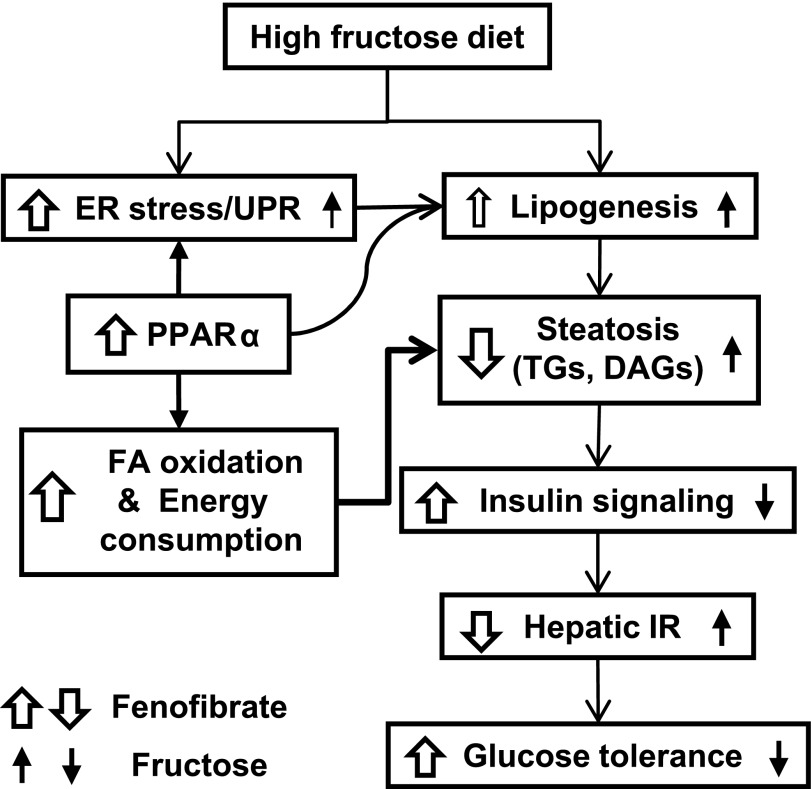
Endoplasmic reticulum (ER) stress is suggested to cause hepatic insulin resistance by increasing de novo lipogenesis (DNL) and directly interfering with insulin signaling through the activation of the c-Jun N-terminal kinase (JNK) and IκB kinase (IKK) pathway. The current study interrogated these two proposed mechanisms in a mouse model of hepatic insulin resistance induced by a high fructose (HFru) diet with the treatment of fenofibrate (FB) 100 mg/kg/day, a peroxisome proliferator-activated receptor α (PPARα) agonist known to reduce lipid accumulation while maintaining elevated DNL in the liver. FB administration completely corrected HFru-induced glucose intolerance, hepatic steatosis, and the impaired hepatic insulin signaling (pAkt and pGSK3β). Of note, both the IRE1/XBP1 and PERK/eIF2α arms of unfolded protein response (UPR) signaling were activated. While retaining the elevated DNL (indicated by the upregulation of SREBP1c, ACC, FAS, and SCD1 and [3H]H2O incorporation into lipids), FB treatment markedly increased fatty acid oxidation (indicated by induction of ACOX1, p-ACC, β-HAD activity, and [14C]palmitate oxidation) and eliminated the accumulation of diacylglycerols (DAGs), which is known to have an impact on insulin signaling. Despite the marked activation of UPR signaling, neither JNK nor IKK appeared to be activated. These findings suggest that lipid accumulation (mainly DAGs), rather than the activation of JNK or IKK, is pivotal for ER stress to cause hepatic insulin resistance. Therefore, by reducing the accumulation of deleterious lipids, activation of PPARα can ameliorate hepatic insulin resistance against increased ER stress.
Figures
Similar articles
-
Differing endoplasmic reticulum stress response to excess lipogenesis versus lipid oversupply in relation to hepatic steatosis and insulin resistance.PLoS One. 2012;7(2):e30816. doi: 10.1371/journal.pone.0030816. Epub 2012 Feb 15. PLoS One. 2012. PMID: 22355328 Free PMC article.
-
Fenofibrate insulates diacylglycerol in lipid droplet/ER and preserves insulin signaling transduction in the liver of high fat fed mice.Biochim Biophys Acta. 2015 Jul;1852(7):1511-9. doi: 10.1016/j.bbadis.2015.04.005. Epub 2015 Apr 21. Biochim Biophys Acta. 2015. PMID: 25906681
-
Sustained activation of PPARα by endogenous ligands increases hepatic fatty acid oxidation and prevents obesity in ob/ob mice.FASEB J. 2012 Feb;26(2):628-38. doi: 10.1096/fj.11-194019. Epub 2011 Oct 18. FASEB J. 2012. PMID: 22009939 Free PMC article.
-
Ectopic lipid accumulation: A potential cause for metabolic disturbances and a contributor to the alteration of kidney function.Biochimie. 2013 Nov;95(11):1971-9. doi: 10.1016/j.biochi.2013.07.017. Epub 2013 Jul 27. Biochimie. 2013. PMID: 23896376 Review.
-
Mechanisms for insulin resistance: common threads and missing links.Cell. 2012 Mar 2;148(5):852-71. doi: 10.1016/j.cell.2012.02.017. Cell. 2012. PMID: 22385956 Free PMC article. Review.
Cited by
-
A Hot Water Extract of Curcuma longa L. Improves Fasting Serum Glucose Levels in Participants with Low-Grade Inflammation: Reanalysis of Data from Two Randomized, Double-Blind, Placebo-Controlled Trials.Nutrients. 2022 Sep 13;14(18):3763. doi: 10.3390/nu14183763. Nutrients. 2022. PMID: 36145139 Free PMC article. Clinical Trial.
-
Identification of matrine as a promising novel drug for hepatic steatosis and glucose intolerance with HSP72 as an upstream target.Br J Pharmacol. 2015 Sep;172(17):4303-18. doi: 10.1111/bph.13209. Epub 2015 Jul 14. Br J Pharmacol. 2015. PMID: 26040411 Free PMC article.
-
Apocynin prevents cigarette smoking-induced loss of skeletal muscle mass and function in mice by preserving proteostatic signalling.Br J Pharmacol. 2021 Aug;178(15):3049-3066. doi: 10.1111/bph.15482. Epub 2021 Jun 8. Br J Pharmacol. 2021. PMID: 33817783 Free PMC article.
-
Integrated physiology and systems biology of PPARα.Mol Metab. 2014 Mar 6;3(4):354-71. doi: 10.1016/j.molmet.2014.02.002. eCollection 2014 Jul. Mol Metab. 2014. PMID: 24944896 Free PMC article. Review.
-
Zerumbone, a Natural Cyclic Sesquiterpene of Zingiber zerumbet Smith, Attenuates Nonalcoholic Fatty Liver Disease in Hamsters Fed on High-Fat Diet.Evid Based Complement Alternat Med. 2013;2013:303061. doi: 10.1155/2013/303061. Epub 2013 Oct 9. Evid Based Complement Alternat Med. 2013. PMID: 24223615 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
