

Cellular aspects of prion replication in vitro
- PMID: 23340381
- PMCID: PMC3564126
- DOI: 10.3390/v5010374
Cellular aspects of prion replication in vitro
Abstract
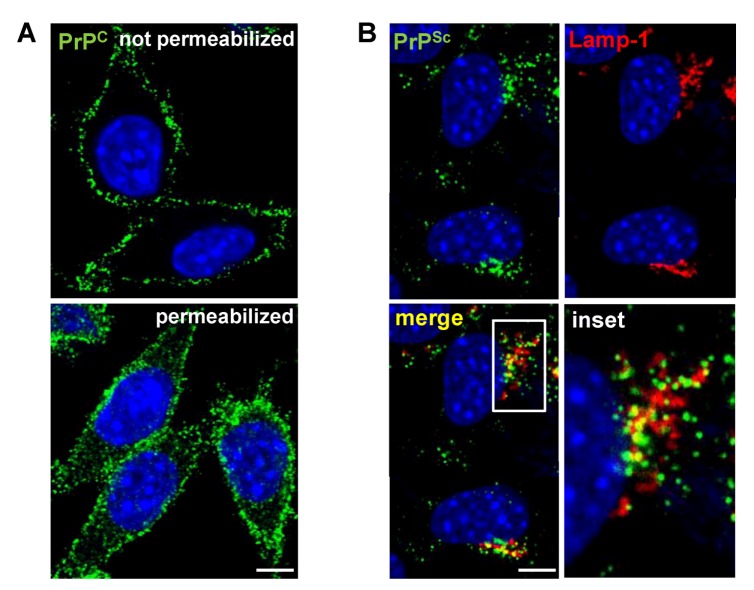
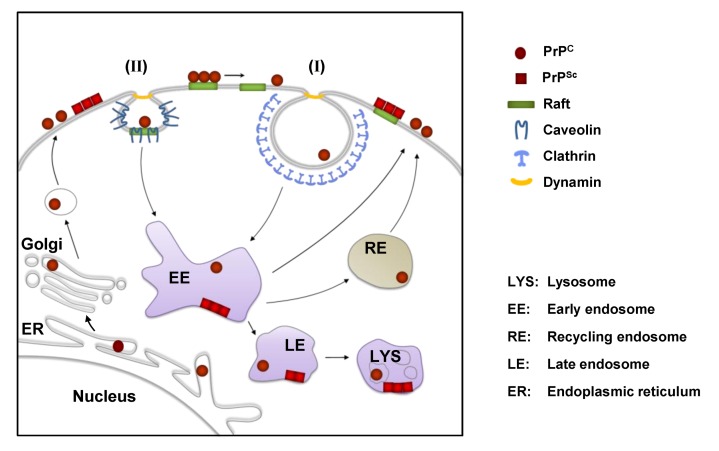
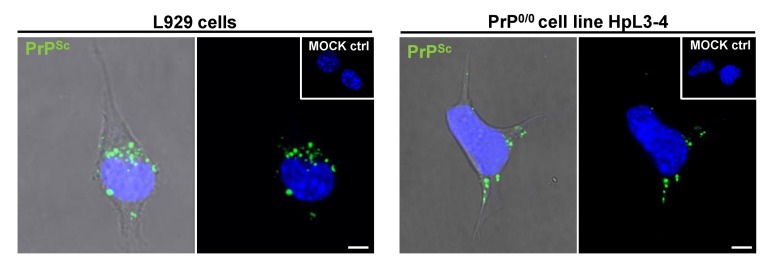
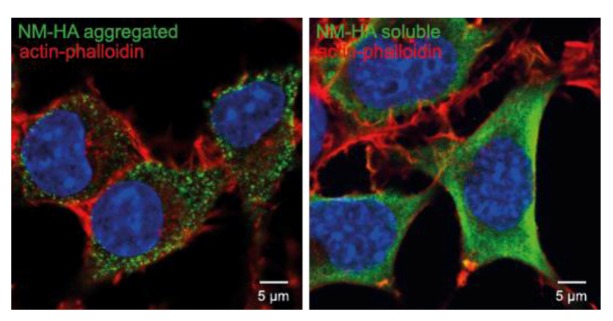
Prion diseases or transmissible spongiform encephalopathies (TSEs) are fatal neurodegenerative disorders in mammals that are caused by unconventional agents predominantly composed of aggregated misfolded prion protein (PrP). Prions self-propagate by recruitment of host-encoded PrP into highly ordered b-sheet rich aggregates. Prion strains differ in their clinical, pathological and biochemical characteristics and are likely to be the consequence of distinct abnormal prion protein conformers that stably replicate their alternate states in the host cell. Understanding prion cell biology is fundamental for identifying potential drug targets for disease intervention. The development of permissive cell culture models has greatly enhanced our knowledge on entry, propagation and dissemination of TSE agents. However, despite extensive research, the precise mechanism of prion infection and potential strain effects remain enigmatic. This review summarizes our current knowledge of the cell biology and propagation of prions derived from cell culture experiments. We discuss recent findings on the trafficking of cellular and pathologic PrP, the potential sites of abnormal prion protein synthesis and potential co-factors involved in prion entry and propagation.
Figures
Similar articles
-
Propagation and Dissemination Strategies of Transmissible Spongiform Encephalopathy Agents in Mammalian Cells.Int J Mol Sci. 2022 Mar 8;23(6):2909. doi: 10.3390/ijms23062909. Int J Mol Sci. 2022. PMID: 35328330 Free PMC article. Review.
-
Transmission and Replication of Prions.Prog Mol Biol Transl Sci. 2017;150:181-201. doi: 10.1016/bs.pmbts.2017.06.014. Epub 2017 Aug 7. Prog Mol Biol Transl Sci. 2017. PMID: 28838661 Review.
-
Protein misfolding cyclic amplification (PMCA): Current status and future directions.Virus Res. 2015 Sep 2;207:47-61. doi: 10.1016/j.virusres.2014.11.007. Epub 2014 Nov 13. Virus Res. 2015. PMID: 25445341 Review.
-
Prion agent diversity and species barrier.Vet Res. 2008 Jul-Aug;39(4):47. doi: 10.1051/vetres:2008024. Epub 2008 Jun 3. Vet Res. 2008. PMID: 18519020 Review.
-
Molecular aspects of disease pathogenesis in the transmissible spongiform encephalopathies.Mol Biotechnol. 2006 May;33(1):71-88. doi: 10.1385/MB:33:1:71. Mol Biotechnol. 2006. PMID: 16691009 Review.
Cited by
-
Caprine PrP variants harboring Asp-146, His-154 and Gln-211 alleles display reduced convertibility upon interaction with pathogenic murine prion protein in scrapie infected cells.Prion. 2016 Sep 2;10(5):391-408. doi: 10.1080/19336896.2016.1199312. Prion. 2016. PMID: 27537339 Free PMC article.
-
Antibody Fragments as Tools for Elucidating Structure-Toxicity Relationships and for Diagnostic/Therapeutic Targeting of Neurotoxic Amyloid Oligomers.Int J Mol Sci. 2020 Nov 24;21(23):8920. doi: 10.3390/ijms21238920. Int J Mol Sci. 2020. PMID: 33255488 Free PMC article. Review.
-
Dual MicroRNA to Cellular Prion Protein Inhibits Propagation of Pathogenic Prion Protein in Cultured Cells.Mol Neurobiol. 2018 Mar;55(3):2384-2396. doi: 10.1007/s12035-017-0495-5. Epub 2017 Mar 29. Mol Neurobiol. 2018. PMID: 28357807
-
Prion strains depend on different endocytic routes for productive infection.Sci Rep. 2017 Jul 31;7(1):6923. doi: 10.1038/s41598-017-07260-2. Sci Rep. 2017. PMID: 28761068 Free PMC article.
-
Oxidative and Inflammatory Events in Prion Diseases: Can They Be Therapeutic Targets?Curr Aging Sci. 2019;11(4):216-225. doi: 10.2174/1874609812666190111100205. Curr Aging Sci. 2019. PMID: 30636622 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
