

Low-resolution structural modeling of protein interactome
- PMID: 23294579
- PMCID: PMC3676717
- DOI: 10.1016/j.sbi.2012.12.003
Low-resolution structural modeling of protein interactome
Abstract
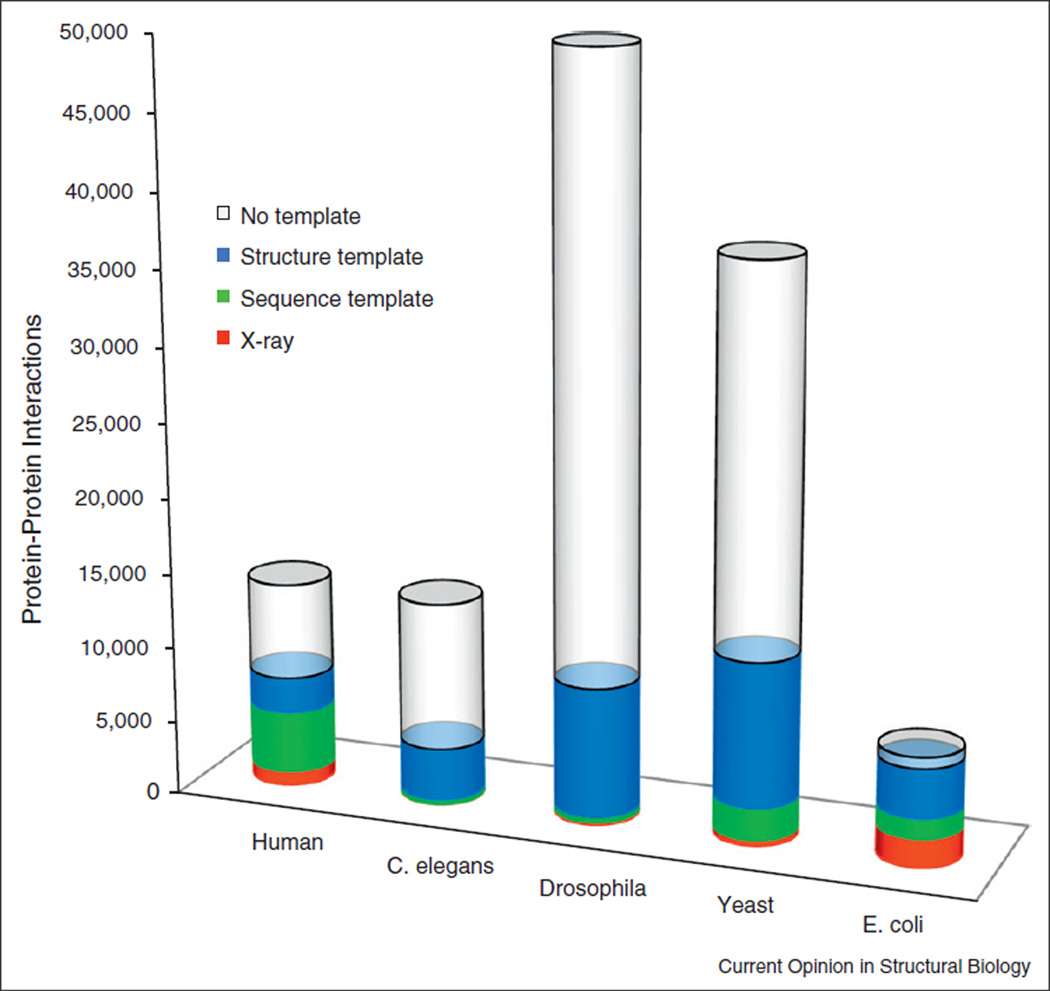
Structural characterization of protein-protein interactions across the broad spectrum of scales is key to our understanding of life at the molecular level. Low-resolution approach to protein interactions is needed for modeling large interaction networks, given the significant level of uncertainties in large biomolecular systems and the high-throughput nature of the task. Since only a fraction of protein structures in interactome are determined experimentally, protein docking approaches are increasingly focusing on modeled proteins. Current rapid advancement of template-based modeling of protein-protein complexes is following a long standing trend in structure prediction of individual proteins. Protein-protein templates are already available for almost all interactions of structurally characterized proteins, and about one third of such templates are likely correct.
Copyright © 2012 Elsevier Ltd. All rights reserved.
Figures
Similar articles
-
Structural quality of unrefined models in protein docking.Proteins. 2017 Jan;85(1):39-45. doi: 10.1002/prot.25188. Epub 2016 Nov 13. Proteins. 2017. PMID: 27756103 Free PMC article.
-
CM2D3: Furnishing the Human Interactome with Structural Models of Protein Complexes Derived by Comparative Modeling and Docking.J Mol Biol. 2023 Jul 15;435(14):168055. doi: 10.1016/j.jmb.2023.168055. Epub 2023 Mar 21. J Mol Biol. 2023. PMID: 36958605
-
Advances in template-based protein docking by utilizing interfaces towards completing structural interactome.Curr Opin Struct Biol. 2015 Dec;35:87-92. doi: 10.1016/j.sbi.2015.10.001. Epub 2015 Nov 9. Curr Opin Struct Biol. 2015. PMID: 26539658 Review.
-
How to choose templates for modeling of protein complexes: Insights from benchmarking template-based docking.Proteins. 2020 Aug;88(8):1070-1081. doi: 10.1002/prot.25875. Epub 2020 Feb 7. Proteins. 2020. PMID: 31994759 Free PMC article.
-
Protein-protein docking: from interaction to interactome.Biophys J. 2014 Oct 21;107(8):1785-1793. doi: 10.1016/j.bpj.2014.08.033. Biophys J. 2014. PMID: 25418159 Free PMC article. Review.
Cited by
-
Modeling complexes of modeled proteins.Proteins. 2017 Mar;85(3):470-478. doi: 10.1002/prot.25183. Epub 2016 Oct 24. Proteins. 2017. PMID: 27701777 Free PMC article.
-
Challenges in protein docking.Curr Opin Struct Biol. 2020 Oct;64:160-165. doi: 10.1016/j.sbi.2020.07.001. Epub 2020 Aug 21. Curr Opin Struct Biol. 2020. PMID: 32836051 Free PMC article. Review.
-
Template-based structure modeling of protein-protein interactions.Curr Opin Struct Biol. 2014 Feb;24:10-23. doi: 10.1016/j.sbi.2013.11.005. Epub 2013 Dec 11. Curr Opin Struct Biol. 2014. PMID: 24721449 Free PMC article. Review.
-
Structural quality of unrefined models in protein docking.Proteins. 2017 Jan;85(1):39-45. doi: 10.1002/prot.25188. Epub 2016 Nov 13. Proteins. 2017. PMID: 27756103 Free PMC article.
-
Simulated unbound structures for benchmarking of protein docking in the DOCKGROUND resource.BMC Bioinformatics. 2015 Jul 31;16(1):243. doi: 10.1186/s12859-015-0672-3. BMC Bioinformatics. 2015. PMID: 26227548 Free PMC article.
References
-
- Vakser IA. Main-chain complementarity in protein–protein recognition. Protein Eng. 1996;9:741–744. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
