

Analysis of alternative cleavage and polyadenylation by 3' region extraction and deep sequencing
- PMID: 23241633
- PMCID: PMC3560312
- DOI: 10.1038/nmeth.2288
Analysis of alternative cleavage and polyadenylation by 3' region extraction and deep sequencing
Abstract
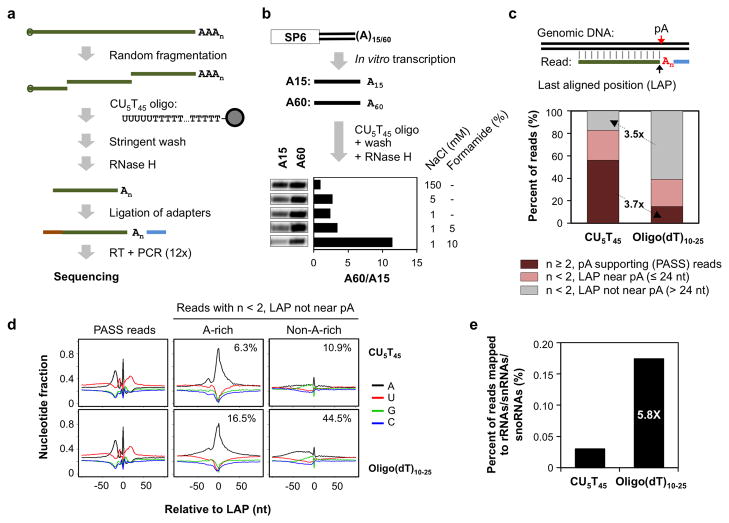
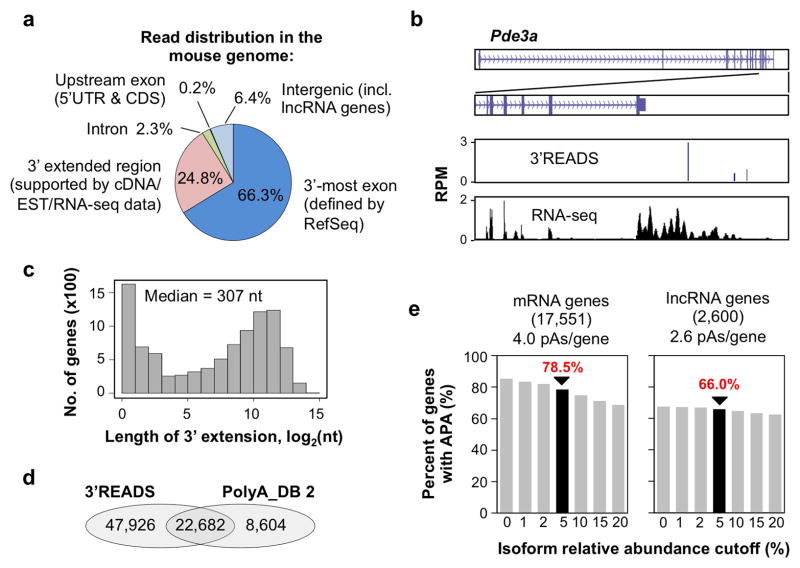
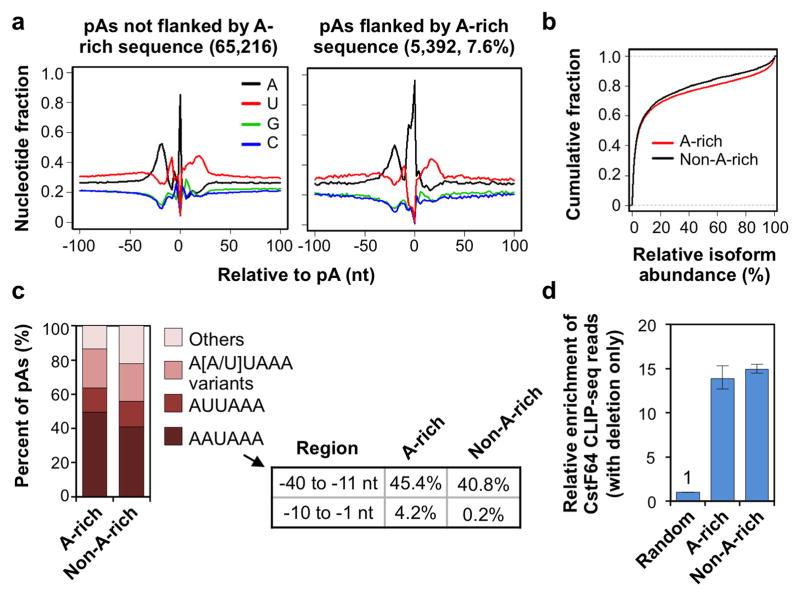
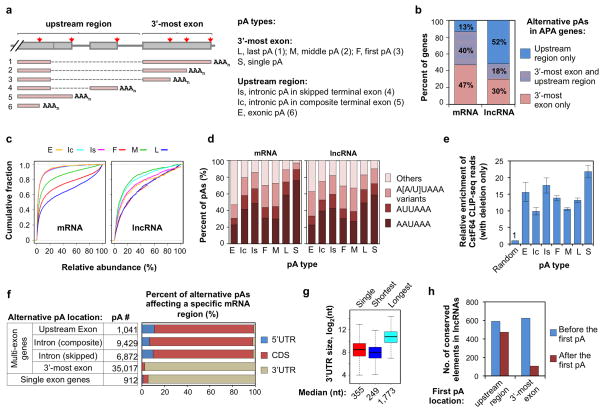
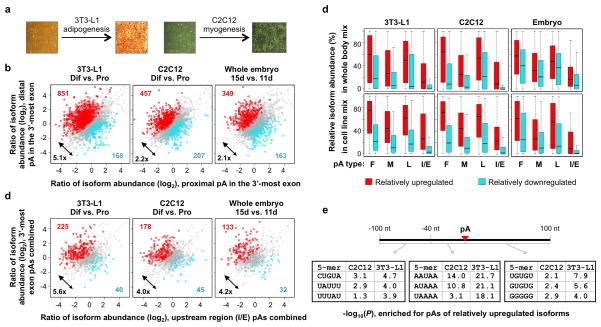
Alternative cleavage and polyadenylation (APA) generates diverse mRNA isoforms. We developed 3' region extraction and deep sequencing (3'READS) to address mispriming issues that commonly plague poly(A) site (pA) identification, and we used the method to comprehensively map pAs in the mouse genome. Thorough annotation of gene 3' ends revealed over 5,000 previously overlooked pAs (∼8% of total) flanked by A-rich sequences, underscoring the necessity of using an accurate tool for pA mapping. About 79% of mRNA genes and 66% of long noncoding RNA genes undergo APA, but these two gene types have distinct usage patterns for pAs in introns and upstream exons. Quantitative analysis of APA isoforms by 3'READS indicated that promoter-distal pAs, regardless of intron or exon locations, become more abundant during embryonic development and cell differentiation and that upregulated isoforms have stronger pAs, suggesting global modulation of the 3' end-processing activity in development and differentiation.
Conflict of interest statement
US Patent application PCT/US2012/052122 based on this work is pending.
Figures
Similar articles
-
3'READS+, a sensitive and accurate method for 3' end sequencing of polyadenylated RNA.RNA. 2016 Oct;22(10):1631-9. doi: 10.1261/rna.057075.116. Epub 2016 Aug 10. RNA. 2016. PMID: 27512124 Free PMC article.
-
Transcription elongation rate has a tissue-specific impact on alternative cleavage and polyadenylation in Drosophila melanogaster.RNA. 2017 Dec;23(12):1807-1816. doi: 10.1261/rna.062661.117. Epub 2017 Aug 29. RNA. 2017. PMID: 28851752 Free PMC article.
-
Accurate mapping of cleavage and polyadenylation sites by 3' region extraction and deep sequencing.Methods Mol Biol. 2014;1125:119-29. doi: 10.1007/978-1-62703-971-0_10. Methods Mol Biol. 2014. PMID: 24590784
-
Implications of polyadenylation in health and disease.Nucleus. 2014;5(6):508-19. doi: 10.4161/nucl.36360. Epub 2014 Oct 31. Nucleus. 2014. PMID: 25484187 Free PMC article. Review.
-
Alternative cleavage and polyadenylation in health and disease.Nat Rev Genet. 2019 Oct;20(10):599-614. doi: 10.1038/s41576-019-0145-z. Epub 2019 Jul 2. Nat Rev Genet. 2019. PMID: 31267064 Review.
Cited by
-
Alternative cleavage and polyadenylation: the long and short of it.Trends Biochem Sci. 2013 Jun;38(6):312-20. doi: 10.1016/j.tibs.2013.03.005. Epub 2013 Apr 27. Trends Biochem Sci. 2013. PMID: 23632313 Free PMC article. Review.
-
Intronic cleavage and polyadenylation regulates gene expression during DNA damage response through U1 snRNA.Cell Discov. 2016 Jun 14;2:16013. doi: 10.1038/celldisc.2016.13. eCollection 2016. Cell Discov. 2016. PMID: 27462460 Free PMC article.
-
Regulating the regulator: a survey of mechanisms from transcription to translation controlling expression of mammalian cell cycle kinase Aurora A.Open Biol. 2022 Sep;12(9):220134. doi: 10.1098/rsob.220134. Epub 2022 Sep 7. Open Biol. 2022. PMID: 36067794 Free PMC article. Review.
-
3'UTR Diversity: Expanding Repertoire of RNA Alterations in Human mRNAs.Mol Cells. 2023 Jan 31;46(1):48-56. doi: 10.14348/molcells.2023.0003. Epub 2023 Jan 20. Mol Cells. 2023. PMID: 36697237 Free PMC article. Review.
-
Alternative polyadenylation regulation in cardiac development and cardiovascular disease.Cardiovasc Res. 2023 Jun 13;119(6):1324-1335. doi: 10.1093/cvr/cvad014. Cardiovasc Res. 2023. PMID: 36657944 Free PMC article. Review.
References
-
- Colgan DF, Manley JL. Mechanism and regulation of mRNA polyadenylation. Genes Dev. 1997;11:2755–2766. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
