

My journey into the world of sphingolipids and sphingolipidoses
- PMID: 23229750
- PMCID: PMC3552047
- DOI: 10.2183/pjab.88.554
My journey into the world of sphingolipids and sphingolipidoses
Abstract
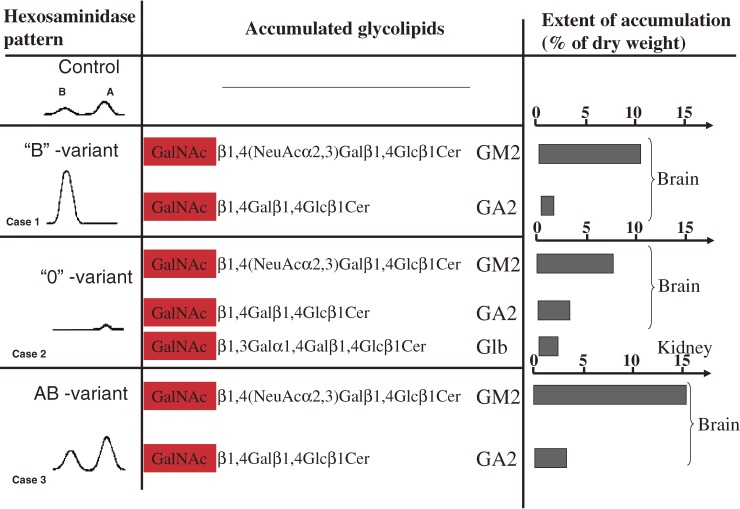
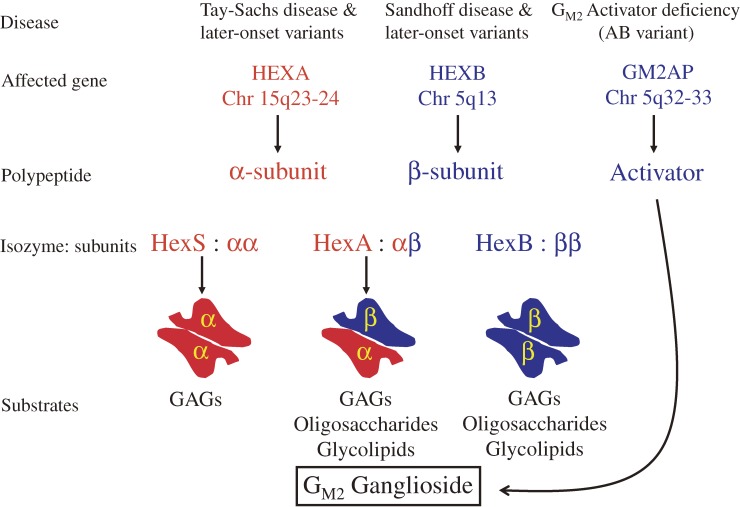
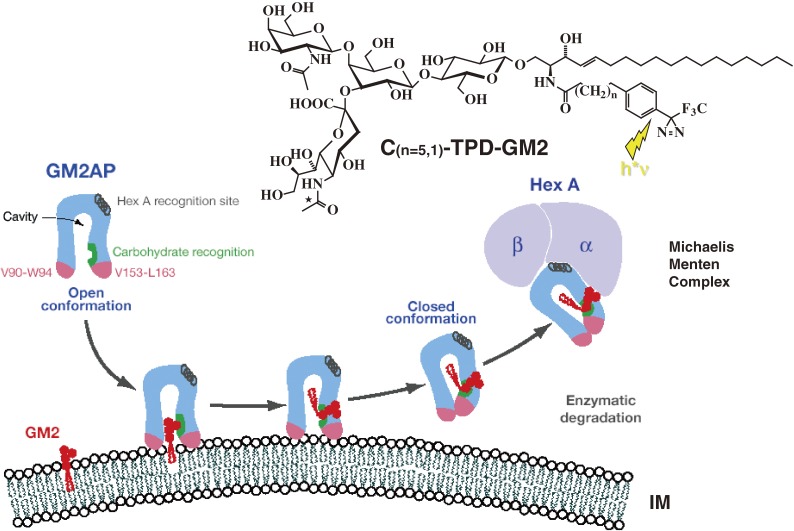
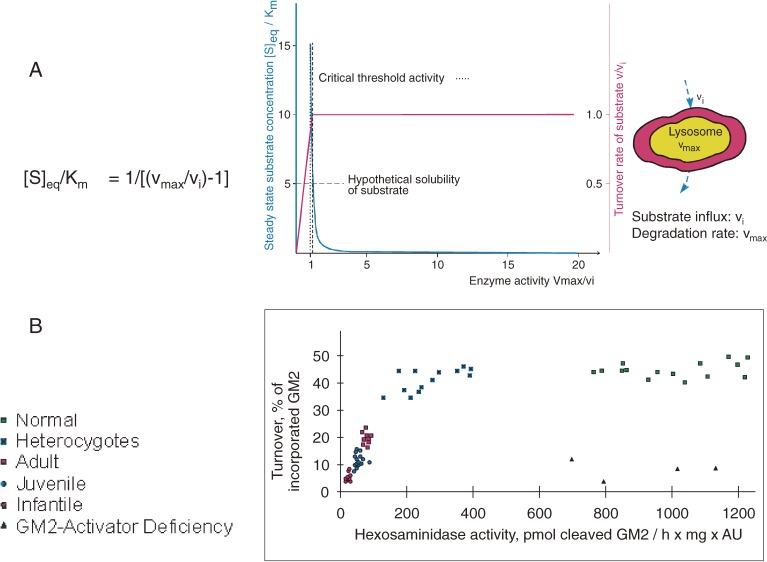
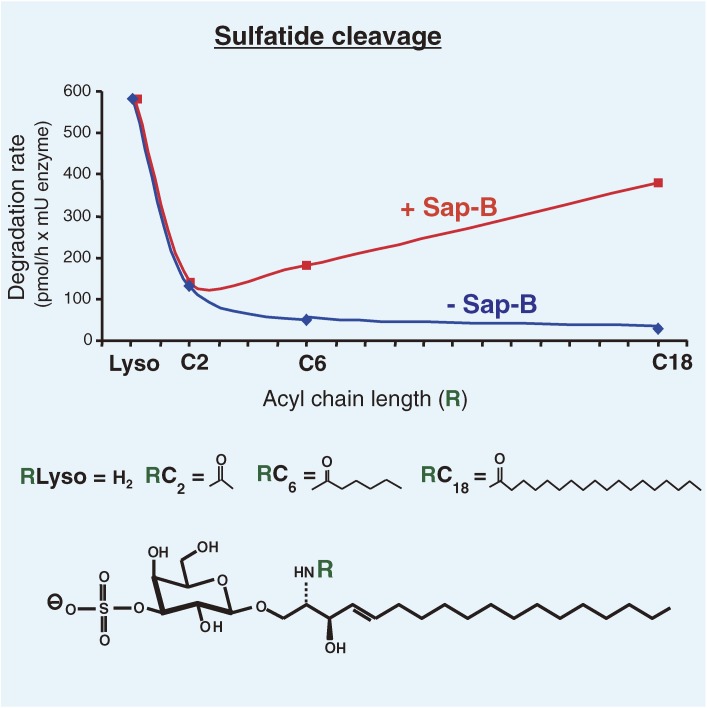
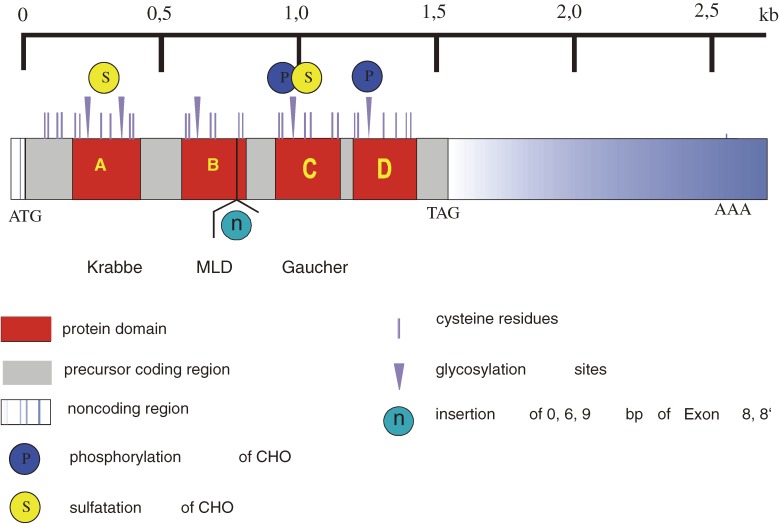
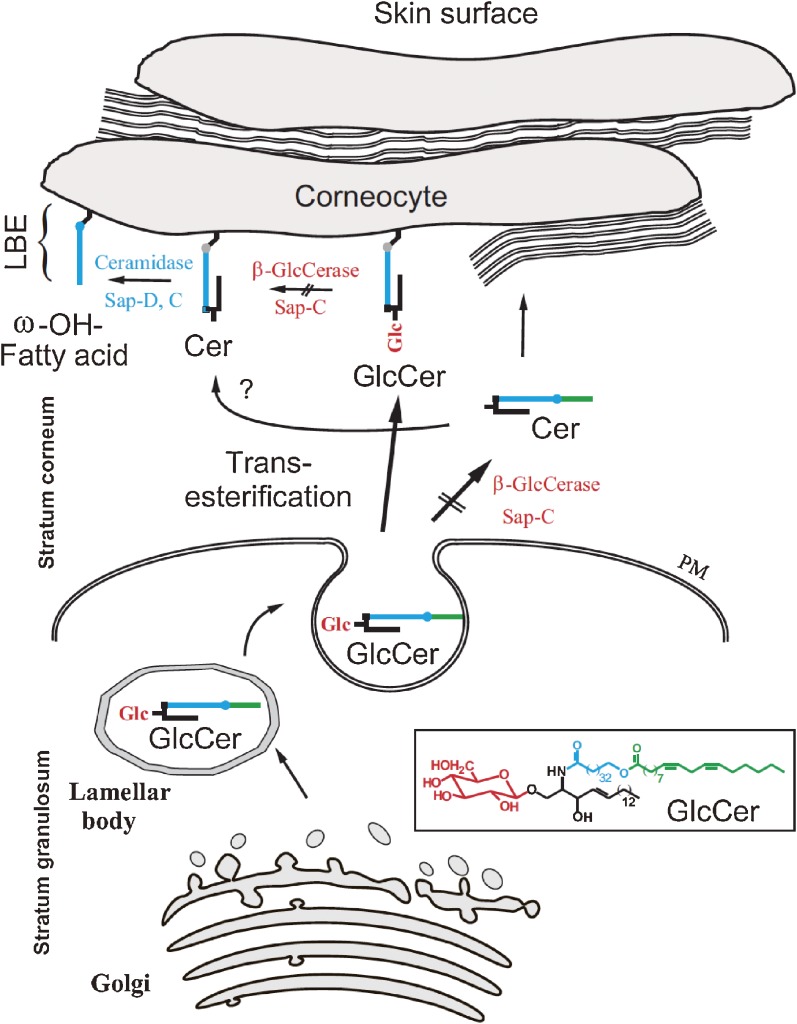
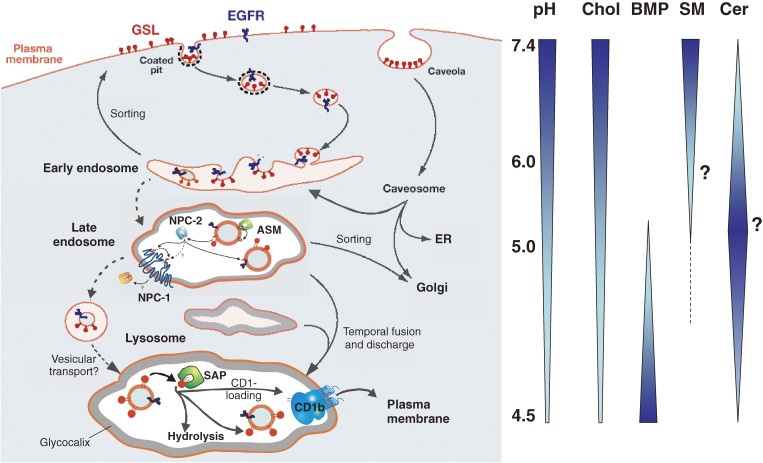
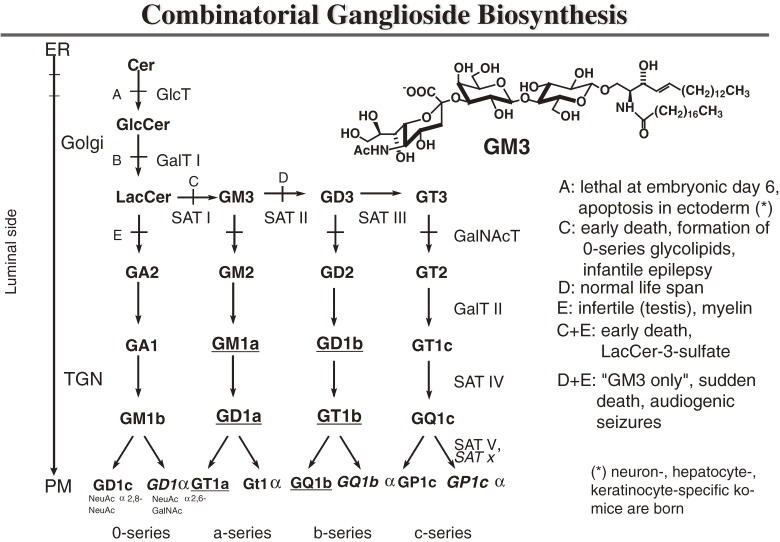
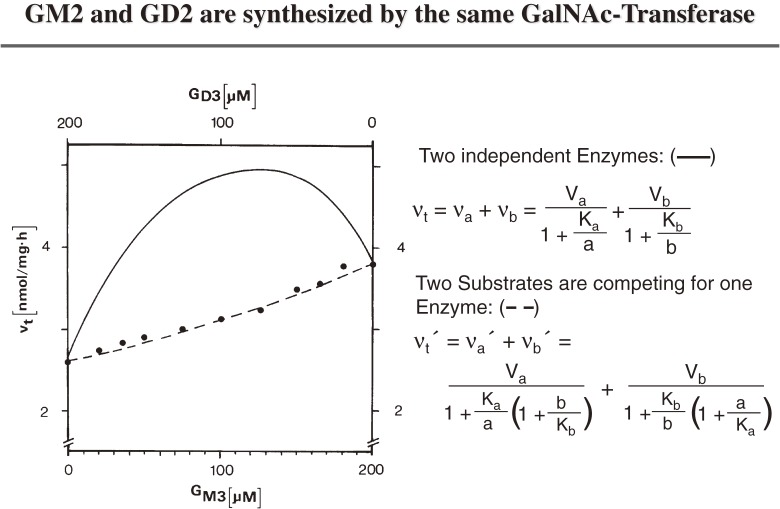
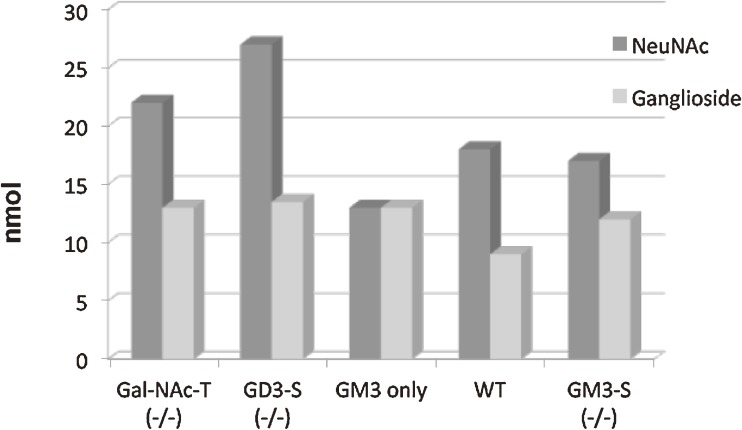
Analysis of lipid storage in postmortem brains of patients with amaurotic idiocy led to the recognition of five lysosomal ganglioside storage diseases and identification of their inherited metabolic blocks. Purification of lysosomal acid sphingomyelinase and ceramidase and analysis of their gene structures were the prerequisites for the clarification of Niemann-Pick and Farber disease. For lipid catabolism, intraendosomal vesicles are formed during the endocytotic pathway. They are subjected to lipid sorting processes and were identified as luminal platforms for cellular lipid and membrane degradation. Lipid binding glycoproteins solubilize lipids from these cholesterol poor membranes and present them to water-soluble hydrolases for digestion. Biosynthesis and intracellular trafficking of lysosomal hydrolases (hexosaminidases, acid sphingomyelinase and ceramidase) and lipid binding and transfer proteins (GM2 activator, saposins) were analyzed to identify the molecular and metabolic basis of several sphingolipidoses. Studies on the biosynthesis of glycosphingolipids yielded the scheme of Combinatorial Ganglioside Biosynthesis involving promiscuous glycosyltransferases. Their defects in mutagenized mice impair brain development and function.
Figures
Similar articles
-
Neuronal sphingolipidoses: Membrane lipids and sphingolipid activator proteins regulate lysosomal sphingolipid catabolism.Biochimie. 2016 Nov;130:146-151. doi: 10.1016/j.biochi.2016.05.004. Epub 2016 May 5. Biochimie. 2016. PMID: 27157270 Review.
-
Metabolic and cellular bases of sphingolipidoses.Biochem Soc Trans. 2013 Dec;41(6):1562-8. doi: 10.1042/BST20130083. Biochem Soc Trans. 2013. PMID: 24256255 Review.
-
Principles of lysosomal membrane digestion: stimulation of sphingolipid degradation by sphingolipid activator proteins and anionic lysosomal lipids.Annu Rev Cell Dev Biol. 2005;21:81-103. doi: 10.1146/annurev.cellbio.21.122303.120013. Annu Rev Cell Dev Biol. 2005. PMID: 16212488 Review.
-
Sphingolipid metabolism. Sphingoid analogs, sphingolipid activator proteins, and the pathology of the cell.Ann N Y Acad Sci. 1998 Jun 19;845:139-51. doi: 10.1111/j.1749-6632.1998.tb09667.x. Ann N Y Acad Sci. 1998. PMID: 9668348 Review.
-
A view on sphingolipids and disease.Chem Phys Lipids. 2011 Sep;164(6):590-606. doi: 10.1016/j.chemphyslip.2011.04.013. Epub 2011 May 6. Chem Phys Lipids. 2011. PMID: 21570958 Review.
Cited by
-
GM2 Gangliosidoses: Clinical Features, Pathophysiological Aspects, and Current Therapies.Int J Mol Sci. 2020 Aug 27;21(17):6213. doi: 10.3390/ijms21176213. Int J Mol Sci. 2020. PMID: 32867370 Free PMC article. Review.
-
Juvenile Tay Sachs Disease Due to Compound Heterozygous Mutation in Hex-A Gene, with Early Sign of Bilateral Tremors.Ann Indian Acad Neurol. 2022 May-Jun;25(3):502-505. doi: 10.4103/aian.aian_577_21. Epub 2022 Jan 5. Ann Indian Acad Neurol. 2022. PMID: 35936646 Free PMC article. No abstract available.
-
Abnormal Sphingolipid World in Inflammation Specific for Lysosomal Storage Diseases and Skin Disorders.Int J Mol Sci. 2018 Jan 15;19(1):247. doi: 10.3390/ijms19010247. Int J Mol Sci. 2018. PMID: 29342918 Free PMC article. Review.
-
LAPTM4B facilitates late endosomal ceramide export to control cell death pathways.Nat Chem Biol. 2015 Oct;11(10):799-806. doi: 10.1038/nchembio.1889. Epub 2015 Aug 17. Nat Chem Biol. 2015. PMID: 26280656
-
Trafficking and Functions of Bioactive Sphingolipids: Lessons from Cells and Model Membranes.Lipid Insights. 2015 Dec 8;8(Suppl 1):11-20. doi: 10.4137/LPI.S31615. eCollection 2015. Lipid Insights. 2015. PMID: 26715852 Free PMC article. Review.
References
-
- Jatzkewitz H. (1958) Zwei Typen von Cerebrosid-Schwefelsäureestern als sog. Prälipoide und Speichersubstanzen bei der Leukodystrophy, Typ Scholz (Metachromatische Form der diffusen Sklerose). Hoppe-Seyler’s Z. Physiol. Chem. 311, 279–282 - PubMed
-
- Jatzkewitz H. (1964) Eine neue Methode zur quantitativen Ultramikrobestimmung der Sphingolipoide aus Gehirn. Hoppe-Seyler’s Z. Physiol. Chem. 336, 25–39 - PubMed
-
- Jatzkewitz H., Mehl E. (1969) Cerebroside-sulphatase and arylsulphatase A deficiency in metachromatic leukodystrophy (ML). J. Neurochem. 16, 19–28 - PubMed
-
- Sandhoff, K. (1965) Die Amaurotische Idiotie des Menschen als Störung im Glykosphingolipidstoffwechsel. Thesis, University of Munich.
-
- Jatzkewitz H., Sandhoff K. (1963) On a biochemically special form of infantile amaurotic idiocy. Biochim. Biophys. Acta 70, 354–356 - PubMed