

Complex DNA repair pathways as possible therapeutic targets to overcome temozolomide resistance in glioblastoma
- PMID: 23227453
- PMCID: PMC3514620
- DOI: 10.3389/fonc.2012.00186
Complex DNA repair pathways as possible therapeutic targets to overcome temozolomide resistance in glioblastoma
Abstract
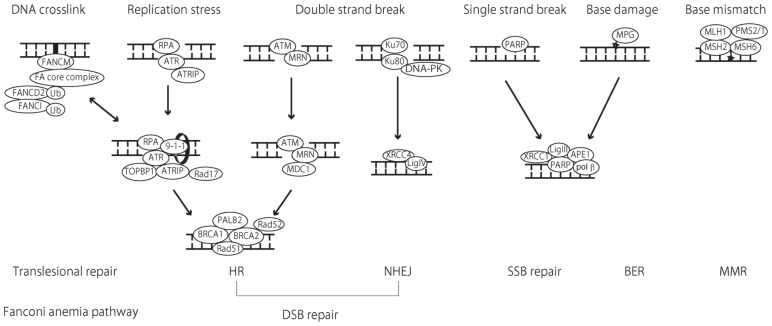
Many conventional chemotherapeutic drugs exert their cytotoxic function by inducing DNA damage in the tumor cell. Therefore, a cell-inherent DNA repair pathway, which reverses the DNA-damaging effect of the cytotoxic drugs, can mediate therapeutic resistance to chemotherapy. The monofunctional DNA-alkylating agent temozolomide (TMZ) is a commonly used chemotherapeutic drug and the gold standard treatment for glioblastoma (GBM). Although the activity of DNA repair protein O6-methylguanine-DNA methyltransferase (MGMT) has been described as the main modulator to determine the sensitivity of GBM to TMZ, a subset of GBM does not respond despite MGMT inactivation, suggesting that another DNA repair mechanism may also modulate the tolerance to TMZ. Considerable interest has focused on MGMT, mismatch repair (MMR), and the base excision repair (BER) pathway in the mechanism of mediating TMZ resistance, but emerging roles for the DNA strand-break repair pathway have been demonstrated. In the first part of this review article, we briefly review the significant role of MGMT, MMR, and the BER pathway in the tolerance to TMZ; in the last part, we review the recent publications that demonstrate possible roles of DNA strand-break repair pathways, such as single-strand break repair and double-strand break repair, as well as the Fanconi anemia pathway in the repair process after alkylating agent-based therapy. It is possible that all of these repair pathways have a potential to modulate the sensitivity to TMZ and aid in overcoming the therapeutic resistance in the clinic.
Keywords: DNA repair; PARP; TMZ; chemoresistance; homologous recombination.
Figures

Similar articles
-
Temozolomide: mechanisms of action, repair and resistance.Curr Mol Pharmacol. 2012 Jan;5(1):102-14. doi: 10.2174/1874467211205010102. Curr Mol Pharmacol. 2012. PMID: 22122467
-
PARP-mediated PARylation of MGMT is critical to promote repair of temozolomide-induced O6-methylguanine DNA damage in glioblastoma.Neuro Oncol. 2021 Jun 1;23(6):920-931. doi: 10.1093/neuonc/noab003. Neuro Oncol. 2021. PMID: 33433610 Free PMC article.
-
Overcoming temozolomide resistance in glioblastoma via dual inhibition of NAD+ biosynthesis and base excision repair.Cancer Res. 2011 Mar 15;71(6):2308-17. doi: 10.1158/0008-5472.CAN-10-3213. Cancer Res. 2011. PMID: 21406402 Free PMC article.
-
Temozolomide resistance in glioblastoma multiforme.Genes Dis. 2016 May 11;3(3):198-210. doi: 10.1016/j.gendis.2016.04.007. eCollection 2016 Sep. Genes Dis. 2016. PMID: 30258889 Free PMC article. Review.
-
Accidental Encounter of Repair Intermediates in Alkylated DNA May Lead to Double-Strand Breaks in Resting Cells.Int J Mol Sci. 2024 Jul 26;25(15):8192. doi: 10.3390/ijms25158192. Int J Mol Sci. 2024. PMID: 39125763 Free PMC article. Review.
Cited by
-
Glesatinib, a c-MET/SMO Dual Inhibitor, Antagonizes P-glycoprotein Mediated Multidrug Resistance in Cancer Cells.Front Oncol. 2019 Apr 25;9:313. doi: 10.3389/fonc.2019.00313. eCollection 2019. Front Oncol. 2019. PMID: 31106148 Free PMC article.
-
Temozolomide and Pituitary Tumors: Current Understanding, Unresolved Issues, and Future Directions.Front Endocrinol (Lausanne). 2018 Jun 15;9:318. doi: 10.3389/fendo.2018.00318. eCollection 2018. Front Endocrinol (Lausanne). 2018. PMID: 29963012 Free PMC article. Review.
-
A stapled peptide antagonist of MDM2 carried by polymeric micelles sensitizes glioblastoma to temozolomide treatment through p53 activation.J Control Release. 2015 Nov 28;218:29-35. doi: 10.1016/j.jconrel.2015.09.061. Epub 2015 Sep 30. J Control Release. 2015. PMID: 26428461 Free PMC article.
-
Cellularly active N-hydroxyurea FEN1 inhibitors block substrate entry to the active site.Nat Chem Biol. 2016 Oct;12(10):815-21. doi: 10.1038/nchembio.2148. Epub 2016 Aug 15. Nat Chem Biol. 2016. PMID: 27526030 Free PMC article.
-
Transcriptomic Profiling of DNA Damage Response in Patient-Derived Glioblastoma Cells before and after Radiation and Temozolomide Treatment.Cells. 2022 Apr 4;11(7):1215. doi: 10.3390/cells11071215. Cells. 2022. PMID: 35406779 Free PMC article.
References
-
- Agnihotri S., Gajadhar A. S., Ternamian C., Gorlia T., Diefes K. L., Mischel P. S., et al. (2012). Alkylpurine-DNA-N-glycosylase confers resistance to temozolomide in xenograft models of glioblastoma multiforme and is associated with poor survival in patients. J. Clin. Invest. 122 253–266 - PMC - PubMed
-
- Al-Ejeh F., Kumar R., Wiegmans A., Lakhani S. R., Brown M. P., Khanna K. K. (2010). Harnessing the complexity of DNA-damage response pathways to improve cancer treatment outcomes. Oncogene 29 6085–6098 - PubMed
-
- Alexander B. M., Pinnell N., Wen P. Y, D’Andrea A. (2012). Targeting DNA repair and the cell cycle in glioblastoma. J. Neurooncol. 107 463–477 - PubMed
-
- Aziz K., Nowsheen S., Pantelias G., Iliakis G., Gorgoulis V. G., Georgakilas A. G. (2012). Targeting DNA damage and repair: embracing the pharmacological era for successful cancer therapy. Pharmacol. Ther. 133 334–350 - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials