

Review
doi: 10.1016/j.virol.2012.09.023.
Snapshots: chromatin control of viral infection
Affiliations
- PMID: 23217624
- PMCID: PMC3531885
- DOI: 10.1016/j.virol.2012.09.023
Item in Clipboard
Review
Snapshots: chromatin control of viral infection
Virology.
.
Abstract
Like their cellular host counterparts, many invading viral pathogens must contend with, modulate, and utilize the host cell's chromatin machinery to promote efficient lytic infection or control persistent-latent states. While not intended to be comprehensive, this review represents a compilation of conceptual snapshots of the dynamic interplay of viruses with the chromatin environment. Contributions focus on chromatin dynamics during infection, viral circumvention of cellular chromatin repression, chromatin organization of large DNA viruses, tethering and persistence, viral interactions with cellular chromatin modulation machinery, and control of viral latency-reactivation cycles.
Published by Elsevier Inc.
Figures
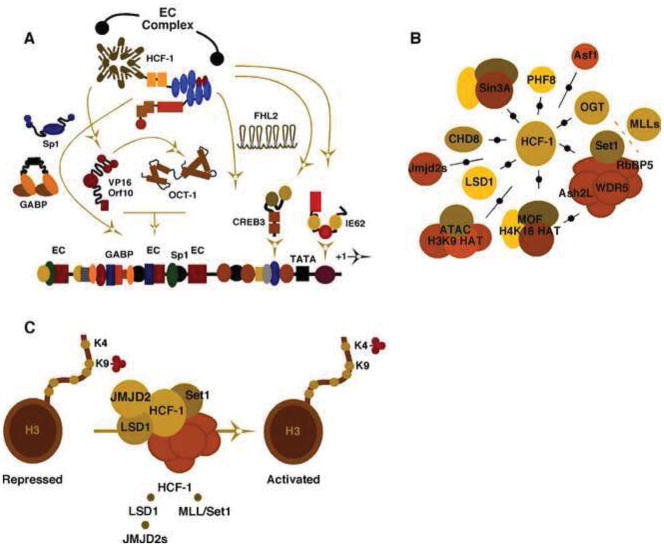
(A). Schematic representation of the complex-combinatorial regulation of the alpha-herpesvirus Immediate Early (IE) genes enhancer-promoters. OCT-1, the viral activator VP16 (HSV) or Orf10 (VZV) form an enhancer core complex (EC complex) with the coactivator HCF-1. Additional cellular and viral activators contribute to the high level transcriptional activation of IE genes and are similarly dependent on HCF-1. (B). Interactions of HCF with multiple chromatin modulation components are illustrated including histone chaperones (Asf1), H3K9 demethylases (PHF8, LSD1, JMJD2s), H3K4 methyltransferases (Set1/MLL complexes), histone acetyltransferases (MOF, ATAC complexes), deacetylases (Sin3A), remodelers (CHD8), and OGT (O-linked N-acetylglucosamine transferase). (C). An HCF-1 complex consisting of histone H3K9 demethylases and H3K4 methyltransferases limits the accumulation of repressive chromatin marks and promotes the installation of activating marks (H3K4-me) for initiation of alpha-herpesvirus IE genes.
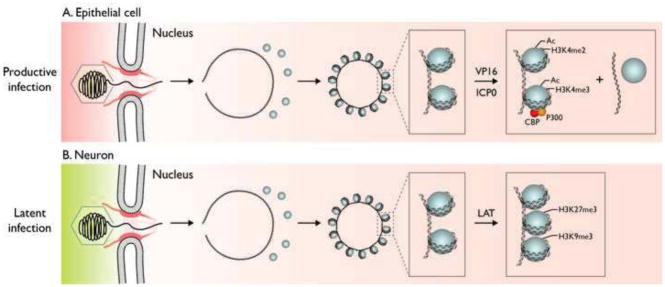
(A). Following infection of epithelial cells, the capsid is transported to the nuclear pore where the viral DNA is released into the nucleus where it rapidly circularizes and becomes associated with histones. VP16 from the virion tegument forms a complex with HCF-1 and Oct1 that binds to viral IE promoters and HCF-1 recruits histone modification enzymes and chromatin remodeling complexes that decrease histone association with viral IE genes and increase euchromatin marks on the remaining associated histones. ICP0 is expressed as an IE protein and it promotes the same processes on the rest of the genome. (B). Following infection of neuronal cells, the capsid is also transported to the nuclear pore where the viral DNA is released into the nucleus where it rapidly circularizes and becomes associated with histones. VP16 cannot be transported into the neuronal nucleus and/or HCF-1 is not localized in the nucleus so viral IE genes are not transcribed efficiently. Instead the latency-associated transcript is expressed and it promotes the association of facultative heterochromatin marks on the viral chromatin (Copyright, Lynne Chang and David Knipe).
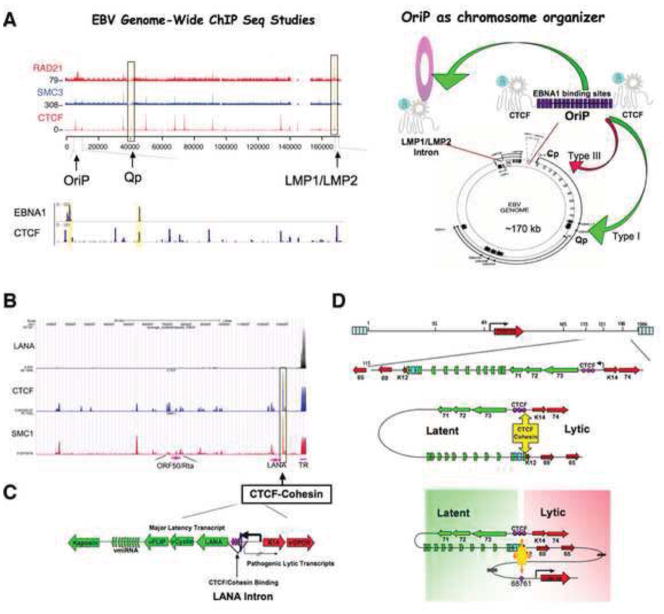
(A). EBV genome-wide ChIP-Seq studies revealed colocalization of CTCF with cohesin subunits Rad21 and SMC3 at several key regulatory sites, including the region at OriP and the LMP1/LMP2 control regions. Viral encoded EBNA1 binds to neighboring sites at the OriP and Qp transcription control regions. OriP functions as a transcriptional enhancer that can physically interact by DNA-looping with the proximal promoters of latency transcripts. (B–C). KSHV genome-wide ChIP-Seq assays reveal CTCF and cohesin co-occupancy at the first intron of the latency transcript encoding LANA-vCyclin-vFLIP multicistronic transcript. (D). Chromatin conformation capture (3C) revealed that this CTCF-cohesin peak mediates DNA-looping with the 3′ end of the latency transcript at K12, as well as a larger loop structure with the lytic cycle immediate early gene promoter control region.

(A) Latent genes (La) are associated with activating histone modifications for their expression. In contrast, immediate early (IE), early (E) and late (L) genes are repressed during latency. IE and E genes possess mainly either acH3/H3K4me3-rich euchromatin or bivalent chromatin (acH3/H3K4me3 and H3K27me3). Heterochromatin characterized by the presence of H3K9me3 and H3K27me3 can also be found on some E genes. Although the majority of late genes are embedded in heterochromatin, some of the late genes have bivalent chromatin. Genome-wide localization of EZH2 H3K27me3 histone methyltransferase of the Polycomb Repressive Complex 2 (PRC2), JMJD2A H3K9me3 histone demethylase and DNA methylation were also mapped onto the KSHV genome during latency. (B). RTA-independent expression of lytic genes. RNAPII binds to the promoter regions of OriLytL, K5, K6 and K7 genes during latency but its activity is blocked by negative transcription elongation factors (DSIF and NELF), which prevents these lytic gene expressions during latency. Positive transcription elongation factor P-TEFb is also recruited to these genes during latency probably in an inactive state. The paused transcription of these lytic genes can be induced either in an RTA-dependent manner as part of the canonical reactivation pathway or in an RTA-independent manner by dissociation of the negative elongation factors. (C). Chromatin control of RTA gene expression. The RTA promoter is controlled by several chromatin modifying enzyme complexes during KSHV infection. This promoter is associated with bivalent chromatin (acH3, H3K4me3 and H3K27me3) during latency that is regulated by PRC2 and different histone deacetylases (HDAC). Chromatin-modifying cellular factors for the induction of the RTA promoter are H3K27me3 histone demethylases (JMJD3 and UTX), H3K4me3 histone methyltransferases (MLL/SET1), histone acetyltransferases (CBP/p300), and chromatin remodeling complex, SWI/SNF2.

The DNA binding domain of E2 binds to specific motifs in the viral genome while other regions interact with host chromatin proteins. This association partitions the viral genome in dividing cells.
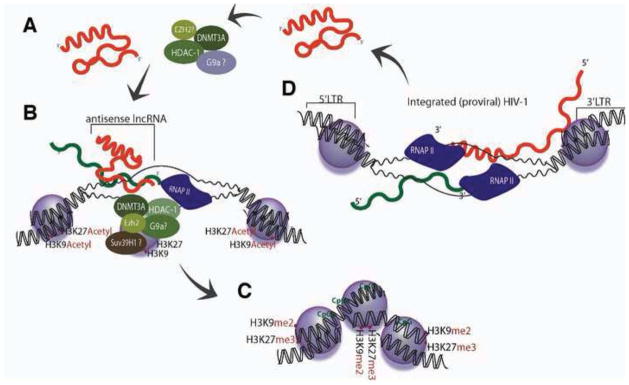
(A) lncRNAs, expressed from pseudogenes or bidirectionally transcribed loci, interact with and recruit various epigenetic remodeling proteins DNA methyltransferase 3A (DNMT3a), Enhancer of Zeste (Ezh2), Histone Deacetylase 1 (HDAC-1) and possibly also G9a. (B) The lncRNA then localizes the epigenetic silencing complex to the homology containing target loci where (C) epigenetic based silencing ensues. (D) HIV-1 expressed antisense lncRNAs, from the 3′ LTR, would also be expected to load into this pathway and guide the epigenetic silencing of integrated forms of HIV-1.
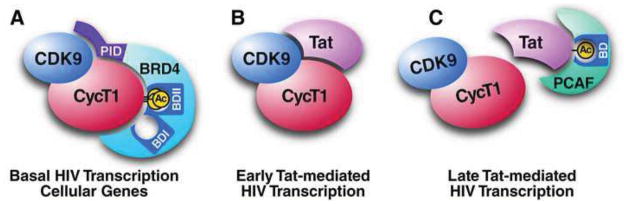
(A) Triacetylated cyclin T1 in P-TEFb interacts with the second bromodomain in Brd4 and regulates basal HIV LTR activity. (B) The Tat protein competes with Brd4 for P-TEFb binding and interacts with nonacetylated cyclin T1 to mediate early steps in Tat-dependent transactivation of the LTR. (C) Tat is acetylated at K50 by p300/KAT3B, a step that dissociates P-TEFb and enables interaction with the bromodomain of PCAF/KAT2B during late steps of Tat-mediated HIV transcription. See text for details.
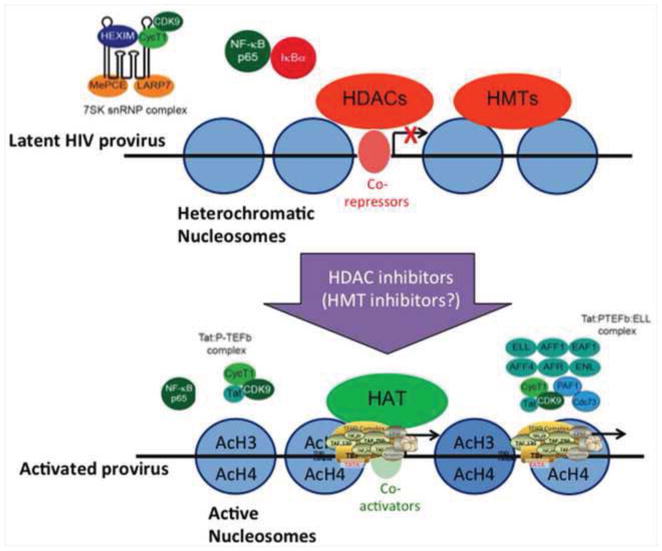
Vorinostat, a Class I-selective HDAC inhibitor, can disrupt HIV latency, in association with mobilization of P-TEFb from the HEXIM complex, and acetylation of proviral nucleosomes, resulting processive transcription mediated by the HIV Tat-P-TEFb-elongation complex. Which of the many effects of HDAC inhibitors is necessary and sufficient for viral induction in vivo has yet to be determined. Methyltransferase inhibitors can synergize with HDAC inhibitors in model systems, but this observation may be difficult to test in vivo given the effects on cellular function that might be induced.
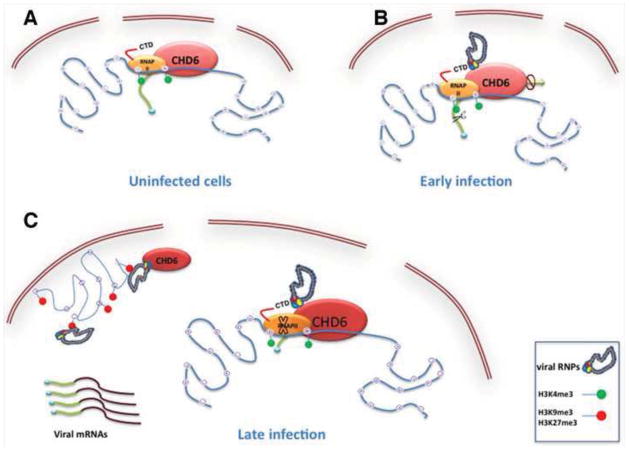
(A). In uninfected cells there is active transcription and CHD6 is together with the RNAP II bound to active chromatin marks (H3K4me3). (B). At early time post-infection, the influenza virus polymerase through its interaction with the RNAP II would couple viral and cellular transcription since viral transcription requires short-capped oligonucleotides from newly synthesized RNAP II transcripts as primers. Once viral transcription is finished, degradation of the RNAP II and inhibition of cellular transcription take place. CHD6 also interacts with the viral polymerase and negatively modulates viral replication. (C). At late time post-infection, viral RNPs are bound to inactive chromatin together with CHD6. The interaction of vRNPs with H3K9me3 and H3K27me3 could cooperate to reorganize cellular chromatin in order to promote the efficient release of newly synthesized vRNPs. At the same time the relocalization of CHD6 to inactive chromatin would inactivate the function of CHD6 since it negatively modulates viral replication.

(A) Presence of nuclear CMV DNA in nucleosomes during productive infection. MRC-5 cells were infected with CMV Towne at a high multiplicity for 0–96 h. Nuclei were reacted with 0.5–75 U micrococcal nuclease (MNase), and purified total DNA was separated in a 1.2% agarose gel stained with ethidium bromide (top). The same DNA samples were subjected to Southern blotting using a 32P-labeled whole genome probe derived from a Towne bacterial artificial chromosome (bottom). Figure provided by Christina Paulus (University of Regensburg). (B) Preferential association of H3K4me2 with replicated CMV chromatin. MRC-5 cells were infected with CMV Towne at a low multiplicity. H3K4me2, nascent DNA, and total DNA were co-stained using appropriate antibodies (rabbit polyclonal antibodies for H3K4me2 and Alexa Fluor 488-coupled goat antibodies for rabbit immunoglobulins), 5-ethynyl-2′-deoxyuridine (EdU)-based “click chemistry”, and 4′,6-diamidino-2-phenylindole (DAPI), respectively. Arrowheads point at a nucleus showing accumulation of H3K4me2 at the site of viral DNA synthesis. Figure provided by Alexandra Asbach-Nitzsche (University of Regensburg). (C) Nucleosome and histone modification dynamics in CMV chromatin. Following release from CMV capsids into the cell nucleus, all or a subset of initially “naked” parental viral genomes undergo rapid de novo nucleosome formation by DNA replication-independent mechanisms resulting in viral chromatin with low nucleosome content. The viral genome-associated nucleosomes are targets of repressive histone modifications that, together with additional nucleosome deposition, may contribute to establishment of viral latent infection. As lytic infection progresses, repressive histone marks are gradually replaced by activating marks including H3K4me2. Newly synthesized viral DNA is subject to replication-coupled nucleosome deposition. Unless a subset of replicated viral genomes stays histone-free, a nucleosome disassembly process must exist to enable DNA packaging and infectious progeny production.
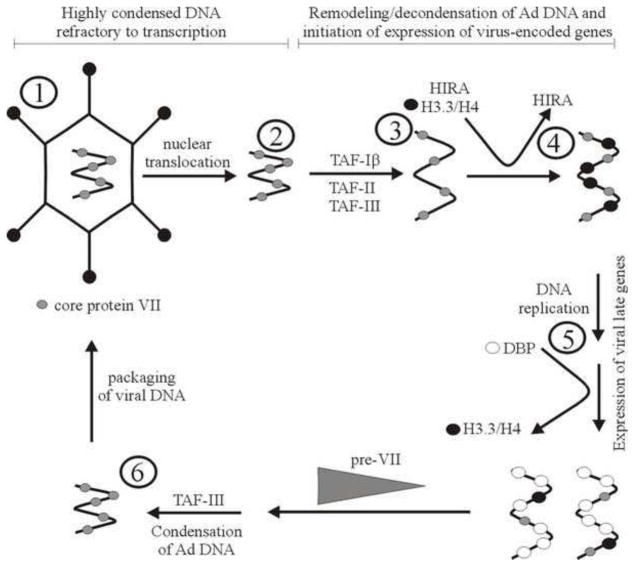
DNA in the Ad capsid is highly condensed with core protein VII (1), along with protein V and mu (not shown). The protein VII-DNA complex transits to the nucleus (2), and undergoes remodeling to decondense the core before transcription of early genes can begin (3). Remodeling may involve loss of at least some VII. At the time when viral gene expression is first detected, histones can be found bound to the viral DNA along with VII. Histone variant H3.3 is preferentially deposited on the viral DNA, through the action of HIRA (4). Onset of viral DNA replication is accompanied by reduced association of the viral DNA with histones, either passively through a reduction in cellular stores of these proteins or actively through exclusion of histones from the viral replication centres by the Ad-encoded DNA binding protein (DBP). Late in infection, the viral DNA must once again associate with pre-protein VII, possibly mediated by TAF-III (6). The viral DNA condensed with pre-protein VII is packaged into the Ad capsid (1). (Adapted from Giberson et al. 2012).
Similar articles
-
Chromatin regulation of virus infection.Trends Microbiol. 2006 Mar;14(3):132-40. doi: 10.1016/j.tim.2006.01.001. Epub 2006 Feb 2. Trends Microbiol. 2006. PMID: 16458005 Review.
-
Epigenetic and epitranscriptomic regulation of viral replication.Nat Rev Microbiol. 2020 Oct;18(10):559-570. doi: 10.1038/s41579-020-0382-3. Epub 2020 Jun 12. Nat Rev Microbiol. 2020. PMID: 32533130 Free PMC article. Review.
-
Viral Interplay with the Host Sumoylation System.Adv Exp Med Biol. 2017;963:359-388. doi: 10.1007/978-3-319-50044-7_21. Adv Exp Med Biol. 2017. PMID: 28197923 Free PMC article. Review.
-
Non-coding RNAs: novel players in chromatin-regulation during viral latency.Curr Opin Virol. 2013 Aug;3(4):387-93. doi: 10.1016/j.coviro.2013.04.001. Epub 2013 May 6. Curr Opin Virol. 2013. PMID: 23660570 Review.
-
The Interplay Between Viral-Derived miRNAs and Host Immunity During Infection.Front Immunol. 2020 Jan 23;10:3079. doi: 10.3389/fimmu.2019.03079. eCollection 2019. Front Immunol. 2020. PMID: 32038626 Free PMC article. Review.
Cited by
-
Navigating Latency-Inducing Viral Infections: Therapeutic Targeting and Nanoparticle Utilization.Biomater Res. 2024 Oct 16;28:0078. doi: 10.34133/bmr.0078. eCollection 2024. Biomater Res. 2024. PMID: 39416703 Free PMC article. Review.
-
Multifunctional Non-Coding RNAs Mediate Latent Infection and Recurrence of Herpes Simplex Viruses.Infect Drug Resist. 2021 Dec 14;14:5335-5349. doi: 10.2147/IDR.S334769. eCollection 2021. Infect Drug Resist. 2021. PMID: 34934329 Free PMC article. Review.
-
Navigating the Host Cell Response during Entry into Sites of Latent Cytomegalovirus Infection.Pathogens. 2018 Mar 16;7(1):30. doi: 10.3390/pathogens7010030. Pathogens. 2018. PMID: 29547547 Free PMC article. Review.
-
Lysine-specific post-translational modifications of proteins in the life cycle of viruses.Cell Cycle. 2019 Sep;18(17):1995-2005. doi: 10.1080/15384101.2019.1639305. Epub 2019 Jul 10. Cell Cycle. 2019. PMID: 31291816 Free PMC article. Review.
-
CCCTC-Binding Factor Acts as a Heterochromatin Barrier on Herpes Simplex Viral Latent Chromatin and Contributes to Poised Latent Infection.mBio. 2018 Feb 6;9(1):e02372-17. doi: 10.1128/mBio.02372-17. mBio. 2018. PMID: 29437926 Free PMC article.
References
-
- Alfonso R, Lutz T, Rodriguez A, Chavez JP, Rodriguez P, Gutierrez S, Nieto A. CHD6 chromatin remodeler is a negative modulator of influenza virus replication that relocates to inactive chromatin upon infection. Cell Microbiol. 2011;13:1894–1906. - PubMed
-
- Archin NM, Liberty AL, Kashuba AD, Choudhary SK, Kuruc JD, Crooks AM, Parker DC, Anderson EM, Kearney MF, Strain MC, Richman DD, Hudgens MG, Bosch RJ, Coffin JM, Eron JJ, Hazuda DJ, Margolis DM. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature. 2012;487:482–485. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 DE017336/DE/NIDCR NIH HHS/United States
- R01CA093606/CA/NCI NIH HHS/United States
- P01AI099783-01/AI/NIAID NIH HHS/United States
- 5-31536/PHS HHS/United States
- R21 AI105909/AI/NIAID NIH HHS/United States
- R01 DE023926/DE/NIDCR NIH HHS/United States
- R56AI096861-01/AI/NIAID NIH HHS/United States
- R01 CA115284/CA/NCI NIH HHS/United States
- R01CA085678/CA/NCI NIH HHS/United States
- R56 AI096861/AI/NIAID NIH HHS/United States
- AI083139/AI/NIAID NIH HHS/United States
- U19 AI096113/AI/NIAID NIH HHS/United States
- R01 AI099081/AI/NIAID NIH HHS/United States
- P01 DE019085/DE/NIDCR NIH HHS/United States
- AI063106/AI/NIAID NIH HHS/United States
- R01 CA117830/CA/NCI NIH HHS/United States
- R01 CA093606/CA/NCI NIH HHS/United States
- P01 AI099783/AI/NIAID NIH HHS/United States
- ZIA AI000711-19/Intramural NIH HHS/United States
- R01 AI063106/AI/NIAID NIH HHS/United States
- R01 DA030156/DA/NIDA NIH HHS/United States
- CAPMC/ CIHR/Canada
- ZIA AI000712-19/Intramural NIH HHS/United States
- P30 AI027763/AI/NIAID NIH HHS/United States
- R01DE017336/DE/NIDCR NIH HHS/United States
- P01DE019085/DE/NIDCR NIH HHS/United States
- R01CA117830/CA/NCI NIH HHS/United States
- R01 CA085678/CA/NCI NIH HHS/United States
- AI099081/AI/NIAID NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
