

Cryo-electron microscopy--a primer for the non-microscopist
- PMID: 23181775
- PMCID: PMC3537914
- DOI: 10.1111/febs.12078
Cryo-electron microscopy--a primer for the non-microscopist
Abstract
Cryo-electron microscopy (cryo-EM) is increasingly becoming a mainstream technology for studying the architecture of cells, viruses and protein assemblies at molecular resolution. Recent developments in microscope design and imaging hardware, paired with enhanced image processing and automation capabilities, are poised to further advance the effectiveness of cryo-EM methods. These developments promise to increase the speed and extent of automation, and to improve the resolutions that may be achieved, making this technology useful to determine a wide variety of biological structures. Additionally, established modalities for structure determination, such as X-ray crystallography and nuclear magnetic resonance spectroscopy, are being routinely integrated with cryo-EM density maps to achieve atomic-resolution models of complex, dynamic molecular assemblies. In this review, which is directed towards readers who are not experts in cryo-EM methodology, we provide an overview of emerging themes in the application of this technology to investigate diverse questions in biology and medicine. We discuss the ways in which these methods are being used to study structures of macromolecular assemblies that range in size from whole cells to small proteins. Finally, we include a description of how the structural information obtained by cryo-EM is deposited and archived in a publicly accessible database.
© 2012 The Authors Journal compilation © 2012 FEBS.
Figures
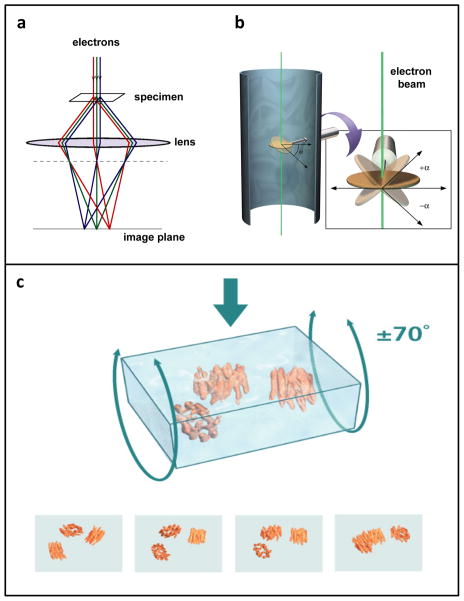
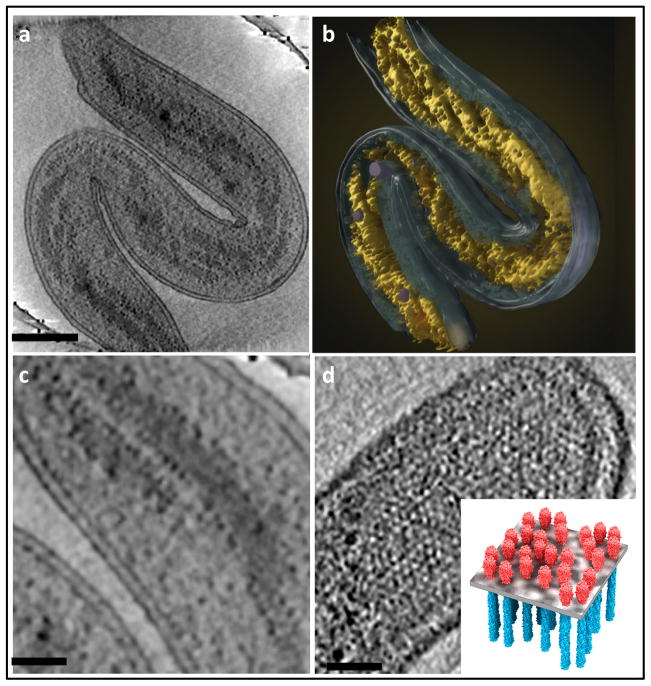
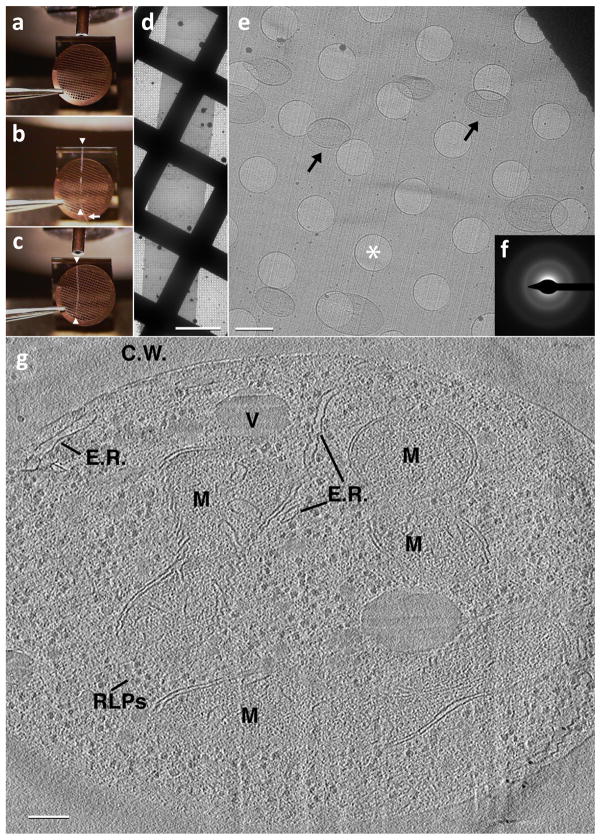
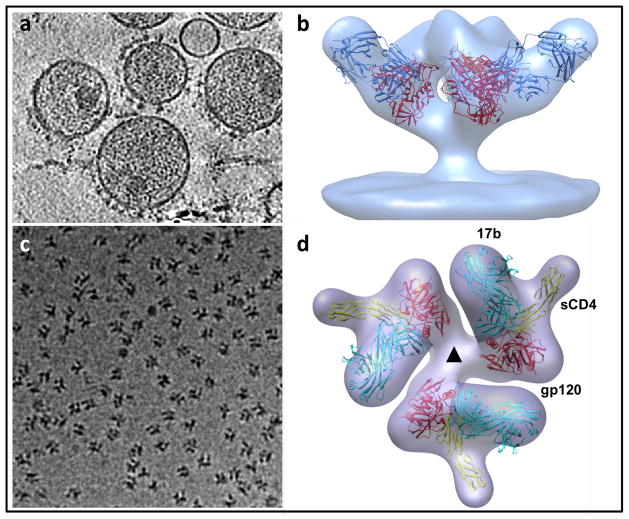
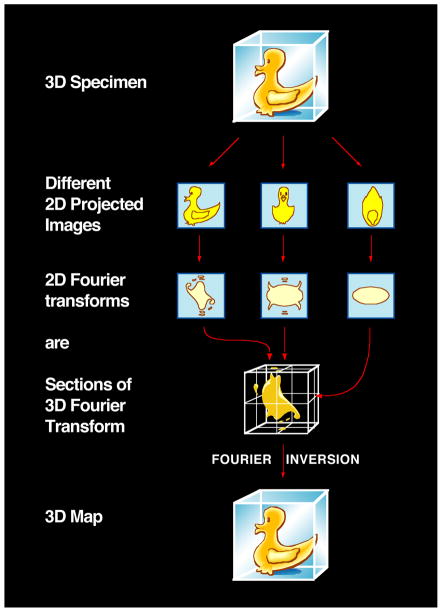
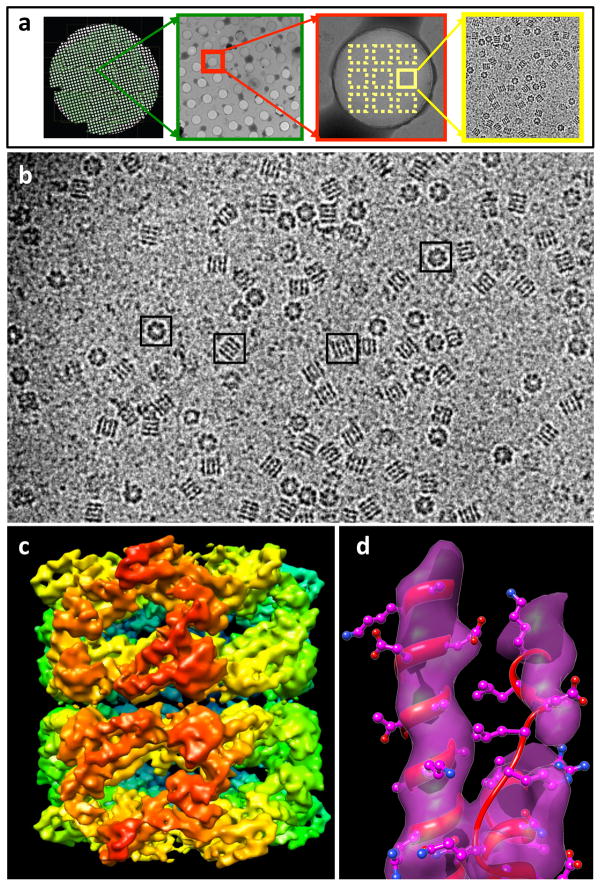
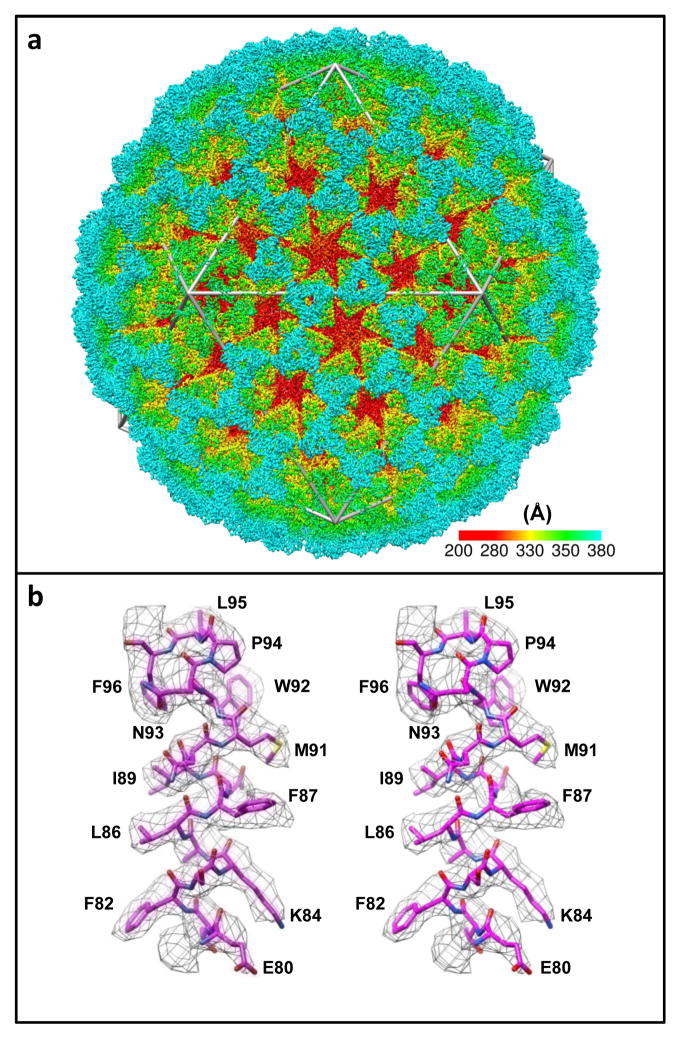

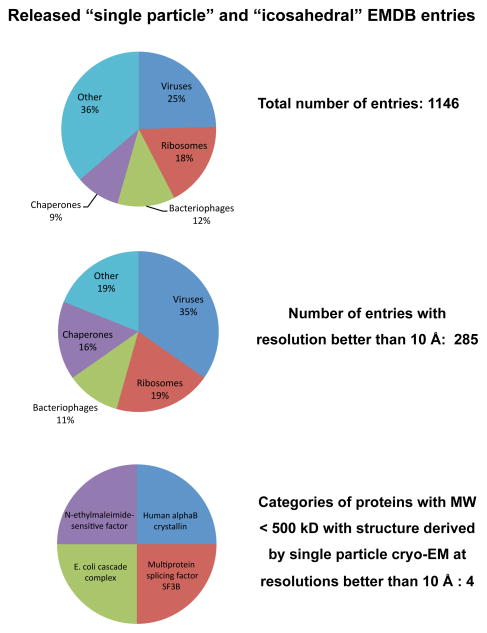
Similar articles
-
Sample preparation of biological macromolecular assemblies for the determination of high-resolution structures by cryo-electron microscopy.Microscopy (Oxf). 2016 Feb;65(1):23-34. doi: 10.1093/jmicro/dfv367. Epub 2015 Dec 15. Microscopy (Oxf). 2016. PMID: 26671943 Review.
-
Cryo electron microscopy to determine the structure of macromolecular complexes.Methods. 2016 Feb 15;95:78-85. doi: 10.1016/j.ymeth.2015.11.023. Epub 2015 Nov 27. Methods. 2016. PMID: 26638773 Free PMC article. Review.
-
Automated structure refinement of macromolecular assemblies from cryo-EM maps using Rosetta.Elife. 2016 Sep 26;5:e17219. doi: 10.7554/eLife.17219. Elife. 2016. PMID: 27669148 Free PMC article.
-
Conventional electron microscopy, cryo-electron microscopy and cryo-electron tomography of viruses.Subcell Biochem. 2013;68:79-115. doi: 10.1007/978-94-007-6552-8_3. Subcell Biochem. 2013. PMID: 23737049 Review.
-
Cryo-electron microscopy and X-ray crystallography: complementary approaches to structural biology and drug discovery.Acta Crystallogr F Struct Biol Commun. 2017 Apr 1;73(Pt 4):174-183. doi: 10.1107/S2053230X17003740. Epub 2017 Mar 29. Acta Crystallogr F Struct Biol Commun. 2017. PMID: 28368275 Free PMC article. Review.
Cited by
-
Cryo-electron microscopy in the study of virus entry and infection.Front Mol Biosci. 2024 Jul 24;11:1429180. doi: 10.3389/fmolb.2024.1429180. eCollection 2024. Front Mol Biosci. 2024. PMID: 39114367 Free PMC article. Review.
-
Membrane Active Peptides and Their Biophysical Characterization.Biomolecules. 2018 Aug 22;8(3):77. doi: 10.3390/biom8030077. Biomolecules. 2018. PMID: 30135402 Free PMC article. Review.
-
Hybrid Vesicle Stability under Sterilisation and Preservation Processes Used in the Manufacture of Medicinal Formulations.Polymers (Basel). 2020 Apr 15;12(4):914. doi: 10.3390/polym12040914. Polymers (Basel). 2020. PMID: 32326448 Free PMC article.
-
Intracellular uranium distribution: Comparison of cryogenic fixation versus chemical fixation methods for SIMS analysis.Microsc Res Tech. 2018 Aug;81(8):855-864. doi: 10.1002/jemt.23047. Epub 2018 May 8. Microsc Res Tech. 2018. PMID: 29737608 Free PMC article.
-
Protein-membrane interactions: sensing and generating curvature.Trends Biochem Sci. 2024 May;49(5):401-416. doi: 10.1016/j.tibs.2024.02.005. Epub 2024 Mar 19. Trends Biochem Sci. 2024. PMID: 38508884 Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
