

Controls of nucleosome positioning in the human genome
- PMID: 23166509
- PMCID: PMC3499251
- DOI: 10.1371/journal.pgen.1003036
Controls of nucleosome positioning in the human genome
Abstract
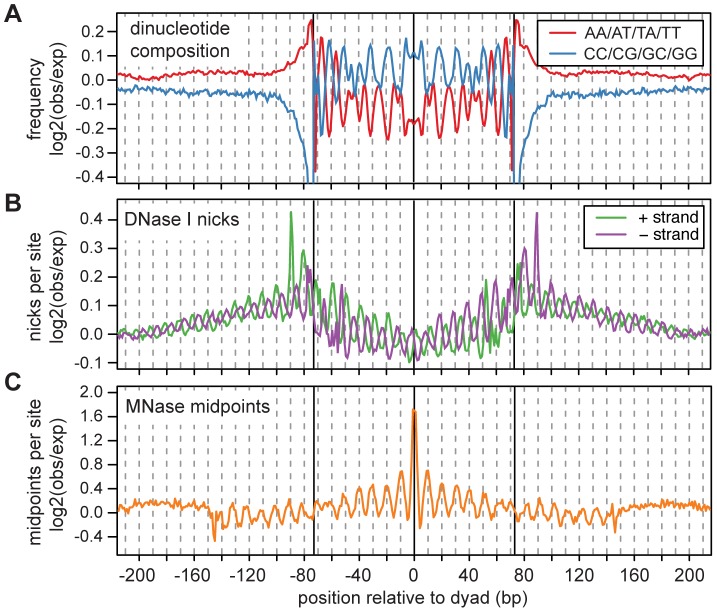
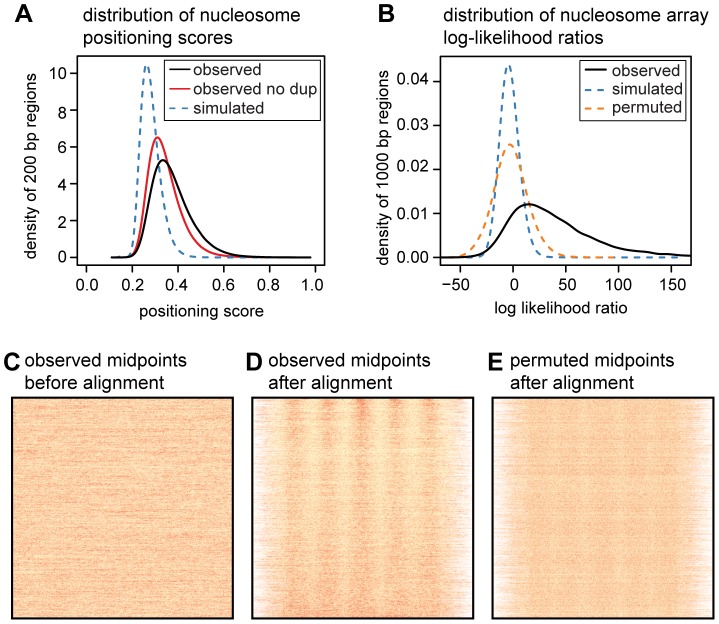
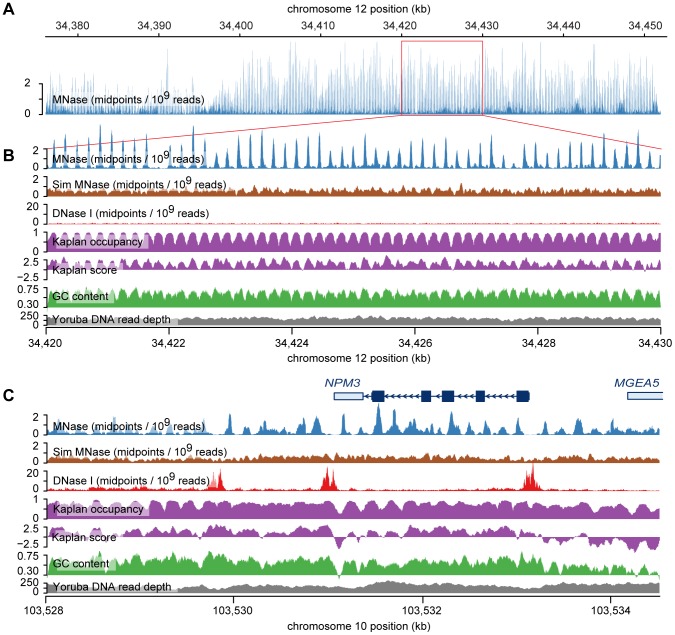
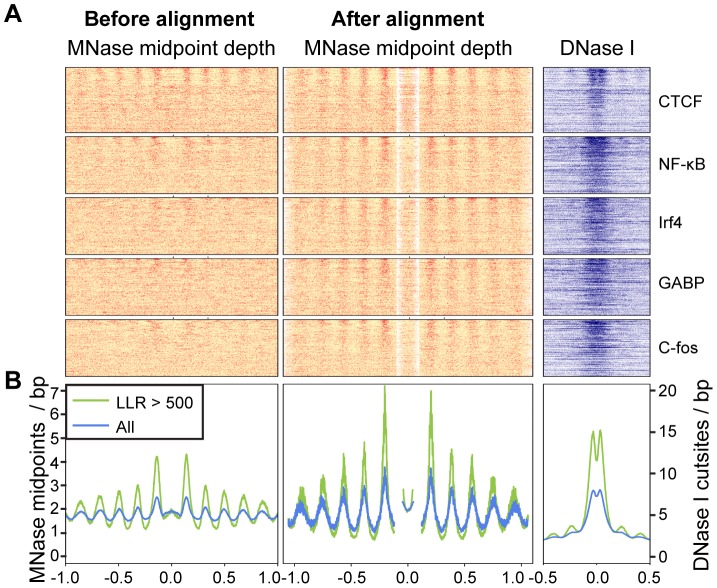
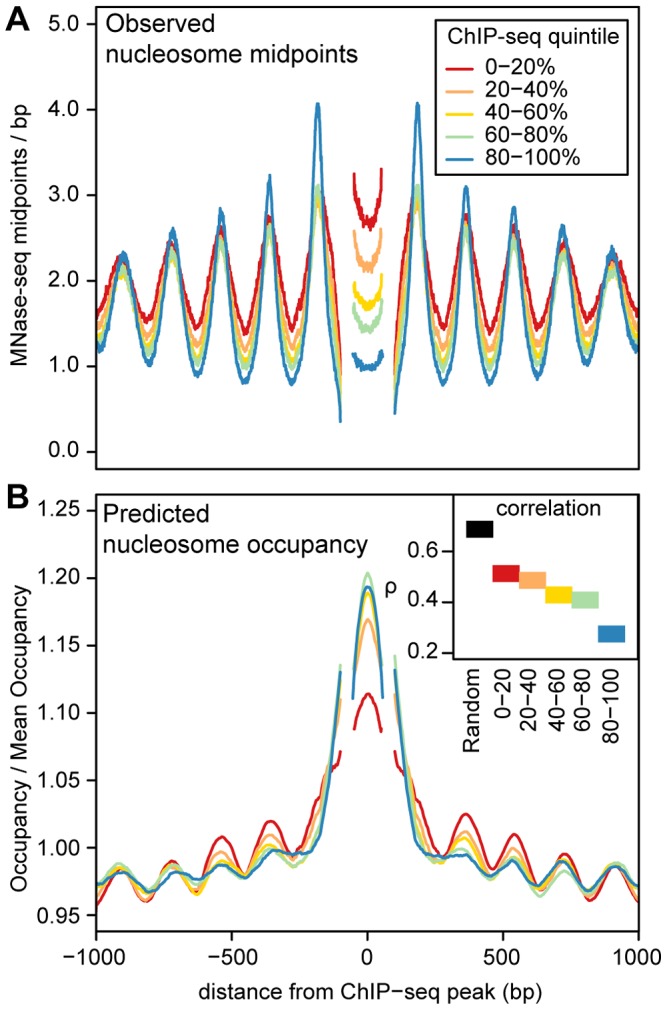
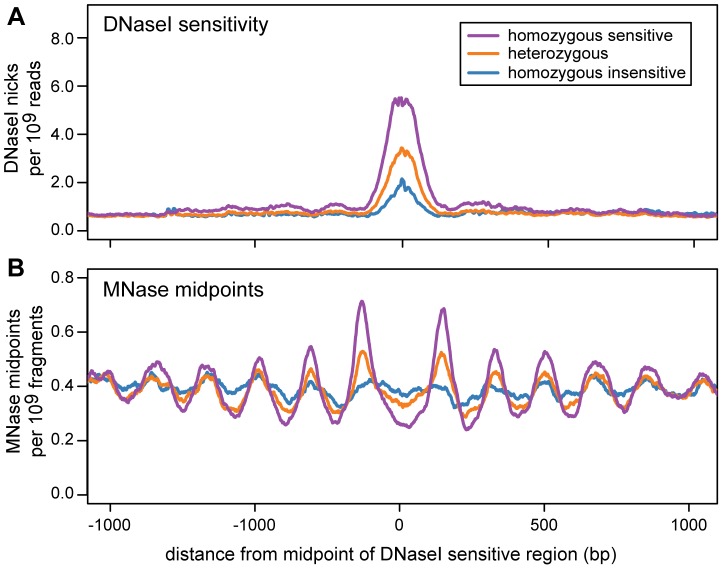
Nucleosomes are important for gene regulation because their arrangement on the genome can control which proteins bind to DNA. Currently, few human nucleosomes are thought to be consistently positioned across cells; however, this has been difficult to assess due to the limited resolution of existing data. We performed paired-end sequencing of micrococcal nuclease-digested chromatin (MNase-seq) from seven lymphoblastoid cell lines and mapped over 3.6 billion MNase-seq fragments to the human genome to create the highest-resolution map of nucleosome occupancy to date in a human cell type. In contrast to previous results, we find that most nucleosomes have more consistent positioning than expected by chance and a substantial fraction (8.7%) of nucleosomes have moderate to strong positioning. In aggregate, nucleosome sequences have 10 bp periodic patterns in dinucleotide frequency and DNase I sensitivity; and, across cells, nucleosomes frequently have translational offsets that are multiples of 10 bp. We estimate that almost half of the genome contains regularly spaced arrays of nucleosomes, which are enriched in active chromatin domains. Single nucleotide polymorphisms that reduce DNase I sensitivity can disrupt the phasing of nucleosome arrays, which indicates that they often result from positioning against a barrier formed by other proteins. However, nucleosome arrays can also be created by DNA sequence alone. The most striking example is an array of over 400 nucleosomes on chromosome 12 that is created by tandem repetition of sequences with strong positioning properties. In summary, a large fraction of nucleosomes are consistently positioned--in some regions because they adopt favored sequence positions, and in other regions because they are forced into specific arrangements by chromatin remodeling or DNA binding proteins.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Kornberg RD (1974) Chromatin structure: a repeating unit of histones and DNA. Science 184: 868–871. - PubMed
-
- Kornberg RD, Lorch Y (1999) Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell 98: 285–294. - PubMed
-
- Kaplan T, Li XY, Sabo PJ, Thomas S, Stamatoyannopoulos JA, et al. (2011) Quantitative Models of the Mechanisms That Control Genome-Wide Patterns of Transcription Factor Binding during Early Drosophila Development. PLoS Genet 7: e1001290 doi:10.1371/journal.pgen.1001290. - DOI - PMC - PubMed
-
- Albert I, Mavrich TN, Tomsho LP, Qi J, Zanton SJ, et al. (2007) Translational and rotational settings of H2A.Z nucleosomes across the Saccharomyces cerevisiae genome. Nature 446: 572–576. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
