

Improving N-terminal protein annotation of Plasmodium species based on signal peptide prediction of orthologous proteins
- PMID: 23153225
- PMCID: PMC3529677
- DOI: 10.1186/1475-2875-11-375
Improving N-terminal protein annotation of Plasmodium species based on signal peptide prediction of orthologous proteins
Abstract
Background: Signal peptide is one of the most important motifs involved in protein trafficking and it ultimately influences protein function. Considering the expected functional conservation among orthologs it was hypothesized that divergence in signal peptides within orthologous groups is mainly due to N-terminal protein sequence misannotation. Thus, discrepancies in signal peptide prediction of orthologous proteins were used to identify misannotated proteins in five Plasmodium species.
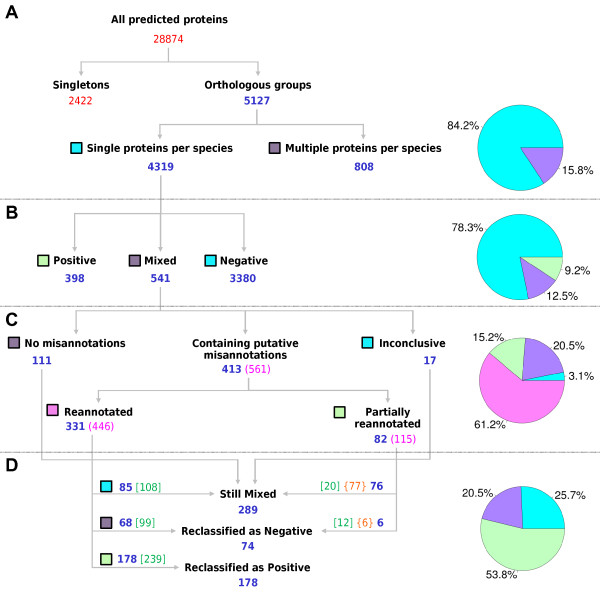
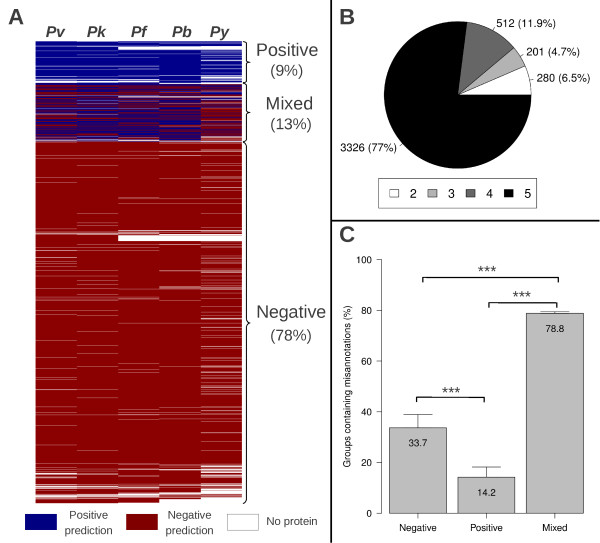
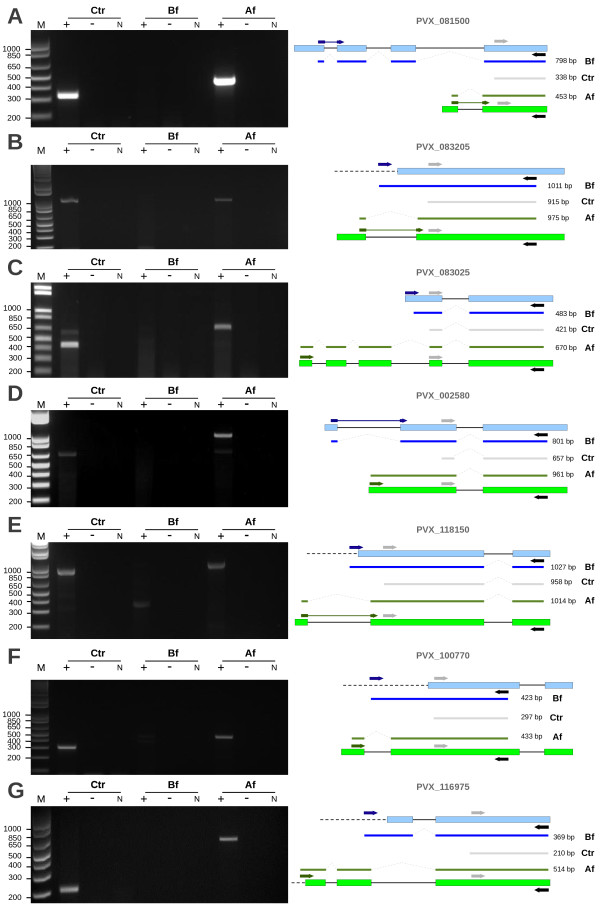
Methods: Signal peptide (SignalP) and orthology (OrthoMCL) were combined in an innovative strategy to identify orthologous groups showing discrepancies in signal peptide prediction among their protein members (Mixed groups). In a comparative analysis, multiple alignments for each of these groups and gene models were visually inspected in search of misannotated proteins and, whenever possible, alternative gene models were proposed. Thresholds for signal peptide prediction parameters were also modified to reduce their impact as a possible source of discrepancy among orthologs. Validation of new gene models was based on RT-PCR (few examples) or on experimental evidence already published (ApiLoc).
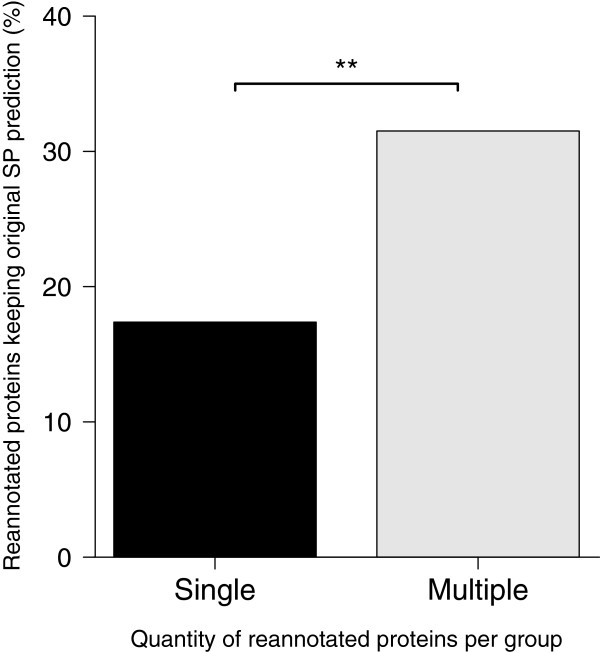
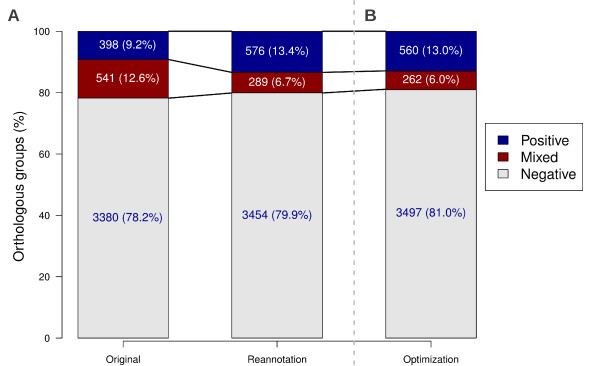
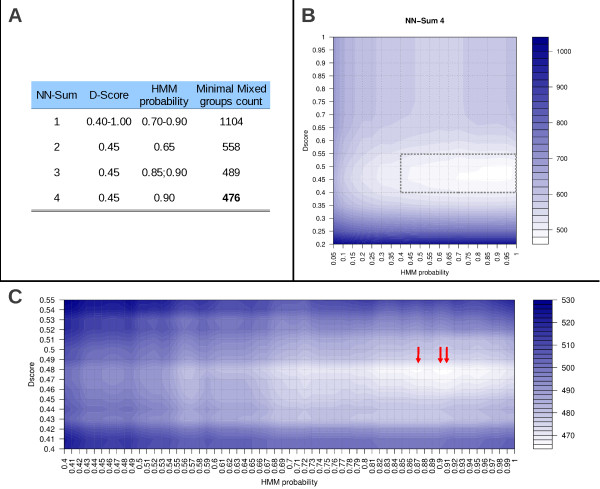
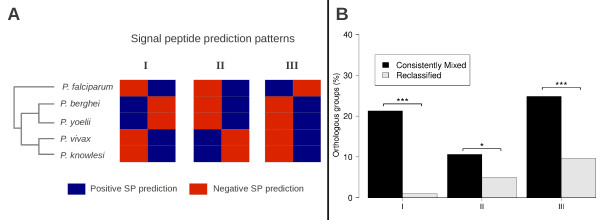
Results: The rate of misannotated proteins was significantly higher in Mixed groups than in Positive or Negative groups, corroborating the proposed hypothesis. A total of 478 proteins were reannotated and change of signal peptide prediction from negative to positive was the most common. Reannotations triggered the conversion of almost 50% of all Mixed groups, which were further reduced by optimization of signal peptide prediction parameters.
Conclusions: The methodological novelty proposed here combining orthology and signal peptide prediction proved to be an effective strategy for the identification of proteins showing wrongly N-terminal annotated sequences, and it might have an important impact in the available data for genome-wide searching of potential vaccine and drug targets and proteins involved in host/parasite interactions, as demonstrated for five Plasmodium species.
Figures
Similar articles
-
In silico identification of genetically attenuated vaccine candidate genes for Plasmodium liver stage.Infect Genet Evol. 2015 Dec;36:72-81. doi: 10.1016/j.meegid.2015.09.002. Epub 2015 Sep 6. Infect Genet Evol. 2015. PMID: 26348884
-
A meta-approach for improving the prediction and the functional annotation of ortholog groups.BMC Genomics. 2014;15 Suppl 6(Suppl 6):S16. doi: 10.1186/1471-2164-15-S6-S16. Epub 2014 Oct 17. BMC Genomics. 2014. PMID: 25573073 Free PMC article.
-
Protein-Coding Gene Families in Prokaryote Genome Comparisons.Methods Mol Biol. 2024;2802:33-55. doi: 10.1007/978-1-0716-3838-5_2. Methods Mol Biol. 2024. PMID: 38819555
-
Proteomic approaches to studying drug targets and resistance in Plasmodium.Curr Drug Targets Infect Disord. 2004 Mar;4(1):41-51. doi: 10.2174/1568005043480989. Curr Drug Targets Infect Disord. 2004. PMID: 15032633 Review.
-
Characteristic features of the SERA multigene family in the malaria parasite.Parasit Vectors. 2020 Apr 6;13(1):170. doi: 10.1186/s13071-020-04044-y. Parasit Vectors. 2020. PMID: 32252804 Free PMC article. Review.
Cited by
-
A putative RNA binding protein from Plasmodium vivax apicoplast.FEBS Open Bio. 2017 Dec 31;8(2):177-188. doi: 10.1002/2211-5463.12351. eCollection 2018 Feb. FEBS Open Bio. 2017. PMID: 29435408 Free PMC article.
-
Automated training for algorithms that learn from genomic data.Biomed Res Int. 2015;2015:234236. doi: 10.1155/2015/234236. Epub 2015 Jan 28. Biomed Res Int. 2015. PMID: 25695053 Free PMC article.
-
Prediction of Signal Peptides in Proteins from Malaria Parasites.Int J Mol Sci. 2018 Nov 22;19(12):3709. doi: 10.3390/ijms19123709. Int J Mol Sci. 2018. PMID: 30469512 Free PMC article.
References
-
- World Health Organization. World Malaria Report. Geneva: WHO Press; 2011.
-
- World Health Organization. World Malaria Report. Geneva: WHO Press; 2010.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
