

RNase L induces autophagy via c-Jun N-terminal kinase and double-stranded RNA-dependent protein kinase signaling pathways
- PMID: 23109342
- PMCID: PMC3527951
- DOI: 10.1074/jbc.M112.399964
RNase L induces autophagy via c-Jun N-terminal kinase and double-stranded RNA-dependent protein kinase signaling pathways
Abstract
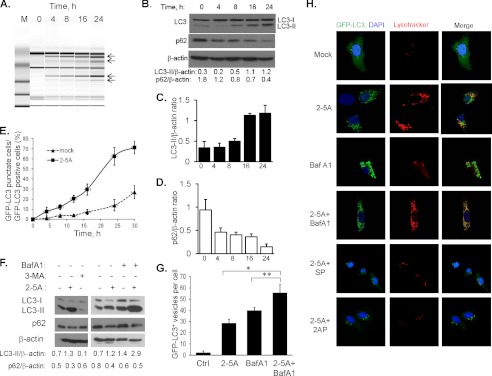
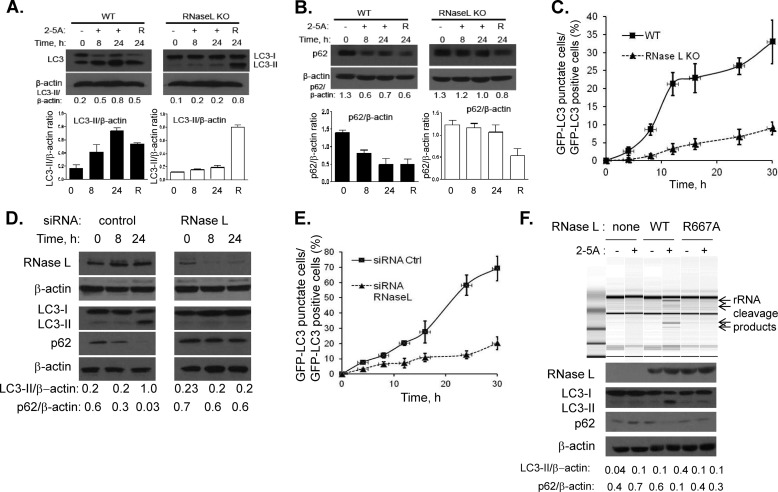
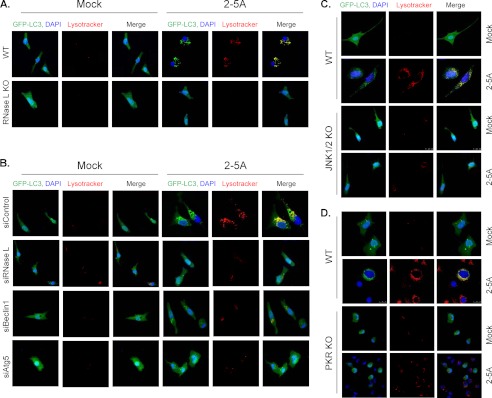
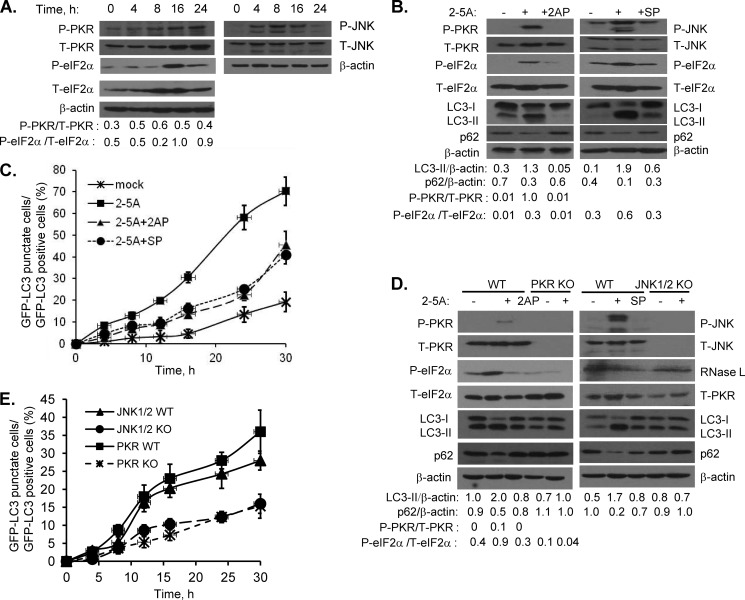
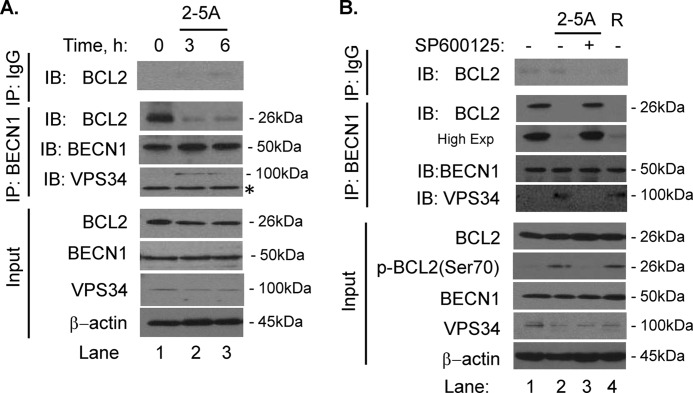
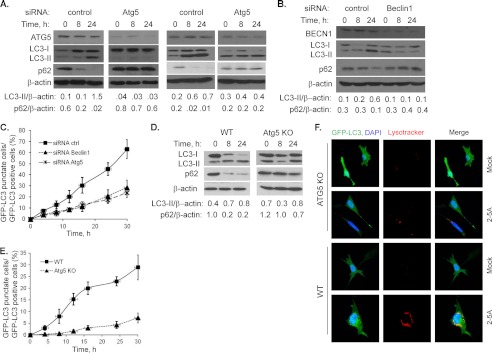
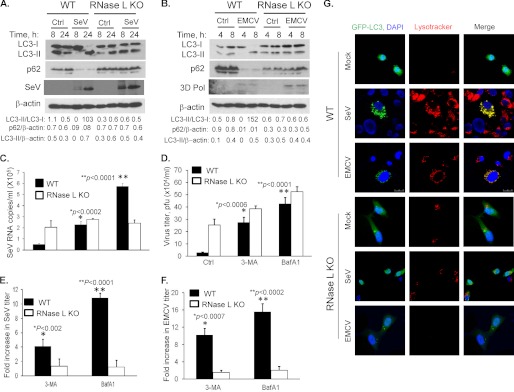
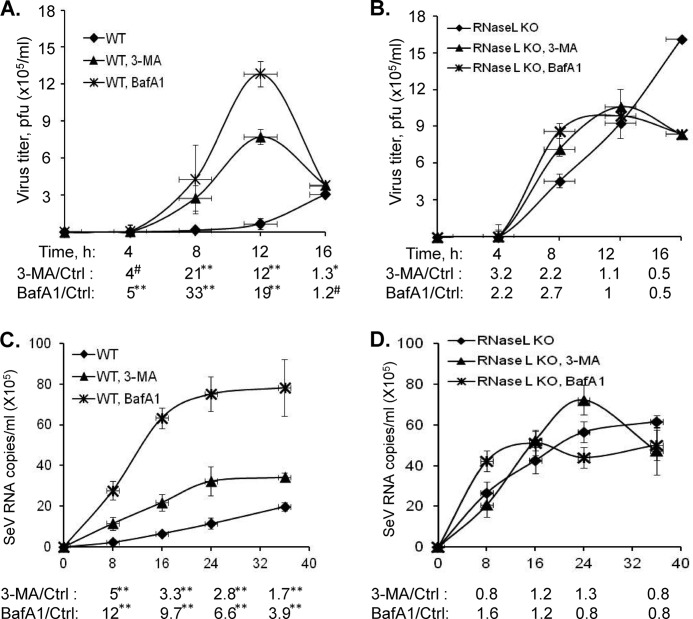
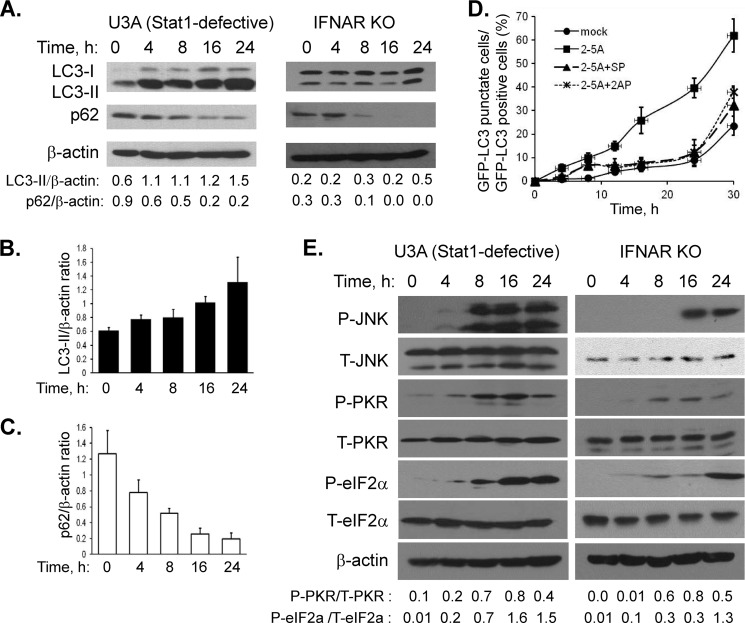
Autophagy is a tightly regulated mechanism that mediates sequestration, degradation, and recycling of cellular proteins, organelles, and pathogens. Several proteins associated with autophagy regulate host responses to viral infections. Ribonuclease L (RNase L) is activated during viral infections and cleaves cellular and viral single-stranded RNAs, including rRNAs in ribosomes. Here we demonstrate that direct activation of RNase L coordinates the activation of c-Jun N-terminal kinase (JNK) and double-stranded RNA-dependent protein kinase (PKR) to induce autophagy with hallmarks as accumulation of autophagic vacuoles, p62(SQSTM1) degradation and conversion of Microtubule-associated Protein Light Chain 3-I (LC3-I) to LC3-II. Accordingly, treatment of cells with pharmacological inhibitors of JNK or PKR and mouse embryonic fibroblasts (MEFs) lacking JNK1/2 or PKR showed reduced autophagy levels. Furthermore, RNase L-induced JNK activity promoted Bcl-2 phosphorylation, disrupted the Beclin1-Bcl-2 complex and stimulated autophagy. Viral infection with Encephalomyocarditis virus (EMCV) or Sendai virus led to higher levels of autophagy in wild-type (WT) MEFs compared with RNase L knock out (KO) MEFs. Inhibition of RNase L-induced autophagy using Bafilomycin A1 or 3-methyladenine suppressed viral growth in initial stages; in later stages autophagy promoted viral replication dampening the antiviral effect. Induction of autophagy by activated RNase L is independent of the paracrine effects of interferon (IFN). Our findings suggest a novel role of RNase L in inducing autophagy affecting the outcomes of viral pathogenesis.
Figures
Similar articles
-
Activation of p38 mitogen-activated protein kinase and c-Jun NH(2)-terminal kinase by double-stranded RNA and encephalomyocarditis virus: involvement of RNase L, protein kinase R, and alternative pathways.Mol Cell Biol. 2000 Jan;20(2):617-27. doi: 10.1128/MCB.20.2.617-627.2000. Mol Cell Biol. 2000. PMID: 10611240 Free PMC article.
-
RNase L triggers autophagy in response to viral infections.J Virol. 2012 Oct;86(20):11311-21. doi: 10.1128/JVI.00270-12. Epub 2012 Aug 8. J Virol. 2012. PMID: 22875977 Free PMC article.
-
JNK-deficiency enhanced oncolytic vaccinia virus replication and blocked activation of double-stranded RNA-dependent protein kinase.Cancer Gene Ther. 2008 Sep;15(9):616-24. doi: 10.1038/cgt.2008.32. Epub 2008 Jun 6. Cancer Gene Ther. 2008. PMID: 18535619 Free PMC article.
-
New insights into the role of RNase L in innate immunity.J Interferon Cytokine Res. 2011 Jan;31(1):49-57. doi: 10.1089/jir.2010.0120. Epub 2010 Dec 29. J Interferon Cytokine Res. 2011. PMID: 21190483 Free PMC article. Review.
-
The Roles of c-Jun N-Terminal Kinase (JNK) in Infectious Diseases.Int J Mol Sci. 2021 Sep 6;22(17):9640. doi: 10.3390/ijms22179640. Int J Mol Sci. 2021. PMID: 34502556 Free PMC article. Review.
Cited by
-
New advances in our understanding of the "unique" RNase L in host pathogen interaction and immune signaling.Cytokine. 2020 Sep;133:153847. doi: 10.1016/j.cyto.2016.08.009. Epub 2016 Aug 29. Cytokine. 2020. PMID: 27595182 Free PMC article. Review.
-
A new mechanism of interferon's antiviral action: Induction of autophagy, essential for paramyxovirus replication, is inhibited by the interferon stimulated gene, TDRD7.PLoS Pathog. 2018 Jan 30;14(1):e1006877. doi: 10.1371/journal.ppat.1006877. eCollection 2018 Jan. PLoS Pathog. 2018. PMID: 29381763 Free PMC article.
-
Epizootic hemorrhagic disease virus induces and benefits from cell stress, autophagy, and apoptosis.J Virol. 2013 Dec;87(24):13397-408. doi: 10.1128/JVI.02116-13. Epub 2013 Oct 2. J Virol. 2013. PMID: 24089565 Free PMC article.
-
Glycyrrhetinic acid induces cytoprotective autophagy via the inositol-requiring enzyme 1α-c-Jun N-terminal kinase cascade in non-small cell lung cancer cells.Oncotarget. 2015 Dec 22;6(41):43911-26. doi: 10.18632/oncotarget.6084. Oncotarget. 2015. PMID: 26549806 Free PMC article.
-
Canonical and Noncanonical Autophagy as Potential Targets for COVID-19.Cells. 2020 Jul 5;9(7):1619. doi: 10.3390/cells9071619. Cells. 2020. PMID: 32635598 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
