

When a domain is not a domain, and why it is important to properly filter proteins in databases: conflicting definitions and fold classification systems for structural domains make filtering of such databases imperative
- PMID: 23108912
- PMCID: PMC3576730
- DOI: 10.1002/bies.201200116
When a domain is not a domain, and why it is important to properly filter proteins in databases: conflicting definitions and fold classification systems for structural domains make filtering of such databases imperative
Abstract
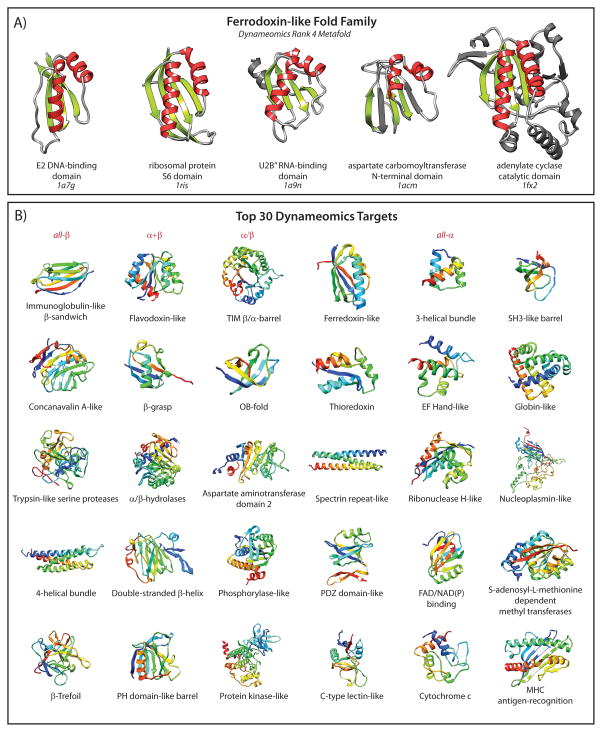
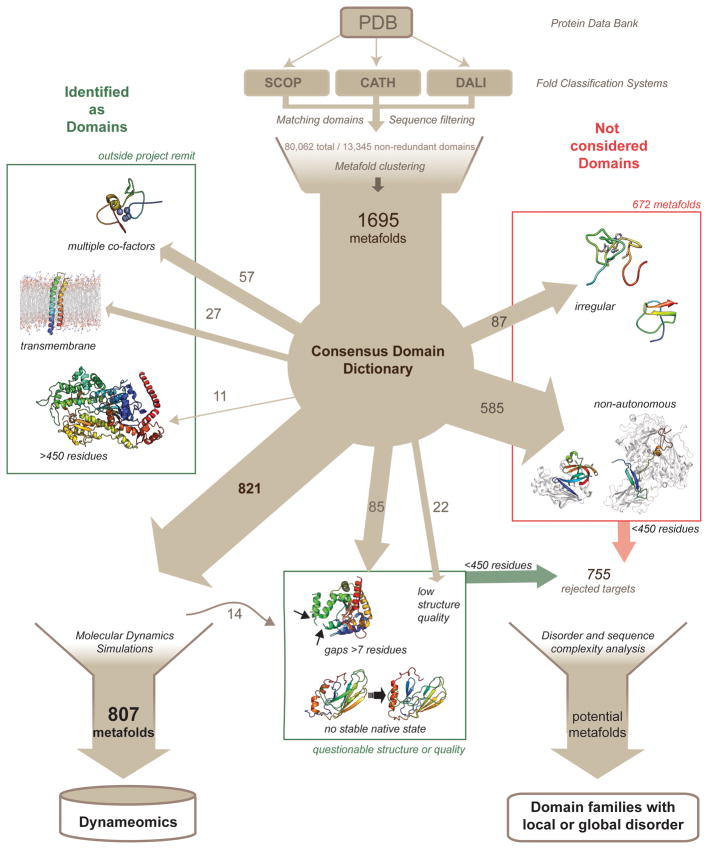
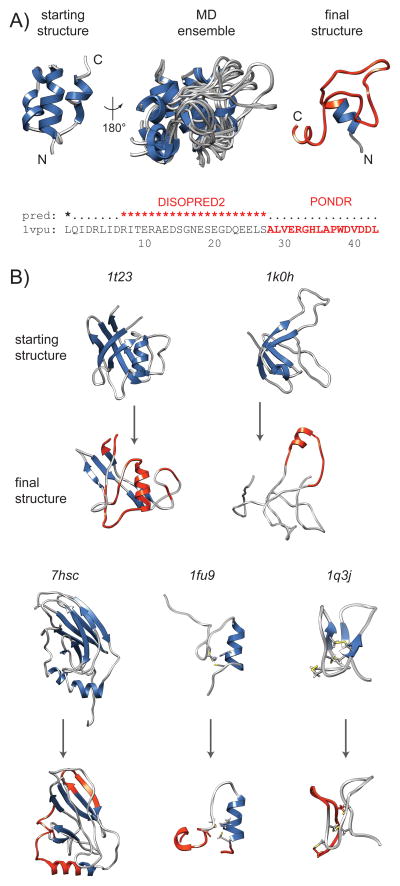
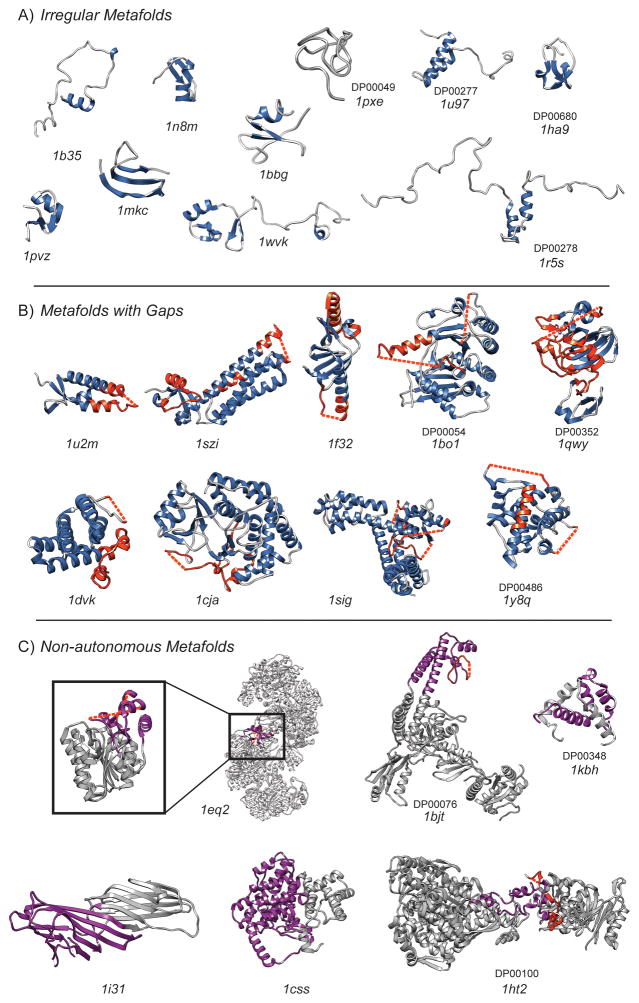
Membership in a protein domain database does not a domain make; a feature we realized when generating a consensus view of protein fold space with our consensus domain dictionary (CDD). This dictionary was used to select representative structures for characterization of the protein dynameome: the Dynameomics initiative. Through this endeavor we rejected a surprising 40% of the 1,695 folds in the CDD as being non-autonomous folding units. Although some of this was due to the challenges of grouping similar fold topologies, the dissonance between the cataloguing and structural qualification of protein domains remains surprising. Another potential factor is previously overlooked intrinsic disorder; predictions suggest that 40% of proteins have either local or global disorder. One thing is clear, filtering a structural database and ensuring a consistent definition for protein domains is crucial, and caution is prescribed when generalizations of globular domains are drawn from unfiltered protein domain datasets.
Copyright © 2012 WILEY Periodicals, Inc.
Figures
Similar articles
-
Generation of a consensus protein domain dictionary.Bioinformatics. 2011 Jan 1;27(1):46-54. doi: 10.1093/bioinformatics/btq625. Epub 2010 Nov 9. Bioinformatics. 2011. PMID: 21068000 Free PMC article.
-
A consensus view of fold space: combining SCOP, CATH, and the Dali Domain Dictionary.Protein Sci. 2003 Oct;12(10):2150-60. doi: 10.1110/ps.0306803. Protein Sci. 2003. PMID: 14500873 Free PMC article.
-
Structural classification of thioredoxin-like fold proteins.Proteins. 2005 Feb 1;58(2):376-88. doi: 10.1002/prot.20329. Proteins. 2005. PMID: 15558583
-
Protein folds and protein folding.Protein Eng Des Sel. 2011 Jan;24(1-2):11-9. doi: 10.1093/protein/gzq096. Epub 2010 Nov 3. Protein Eng Des Sel. 2011. PMID: 21051320 Free PMC article. Review.
-
Overview of protein structural and functional folds.Curr Protoc Protein Sci. 2004 May;Chapter 17(1):Unit 17.1. doi: 10.1002/0471140864.ps1701s35. Curr Protoc Protein Sci. 2004. PMID: 18429251 Free PMC article. Review.
Cited by
-
Protein domain definition should allow for conditional disorder.Protein Sci. 2013 Nov;22(11):1502-18. doi: 10.1002/pro.2336. Epub 2013 Sep 20. Protein Sci. 2013. PMID: 23963781 Free PMC article.
-
Shared unfolding pathways of unrelated immunoglobulin-like β-sandwich proteins.Protein Eng Des Sel. 2019 Dec 31;32(7):331-345. doi: 10.1093/protein/gzz040. Protein Eng Des Sel. 2019. PMID: 31868211 Free PMC article.
-
Conserved patterns and interactions in the unfolding transition state across SH3 domain structural homologues.Protein Sci. 2021 Feb;30(2):391-407. doi: 10.1002/pro.3998. Epub 2020 Nov 26. Protein Sci. 2021. PMID: 33190305 Free PMC article.
-
Protein Fold Usages in Ribosomes: Another Glance to the Past.Int J Mol Sci. 2024 Aug 13;25(16):8806. doi: 10.3390/ijms25168806. Int J Mol Sci. 2024. PMID: 39201491 Free PMC article.
-
New Dynamic Rotamer Libraries: Data-Driven Analysis of Side-Chain Conformational Propensities.Structure. 2016 Jan 5;24(1):187-199. doi: 10.1016/j.str.2015.10.017. Structure. 2016. PMID: 26745530 Free PMC article.
References
-
- Kendrew J, Bodo G, Dintzis H, Parrish R, et al. A three-dimensional model of the myoglobin molecule obtained by X-ray analysis. Nature. 1958;181:662–6. - PubMed
-
- Perutz MF, Rossmann MG, Cullis AF, Muirhead H, et al. Structure of haemoglobin: a three-dimensional Fourier synthesis at 5.5-Å resolution, obtained by X-ray analysis. Nature. 1960;185:416–22. - PubMed
-
- Levitt M, Chothia C. Structural patterns in globular proteins. Nature. 1976;261:552–8. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
