

Bone erosion in rheumatoid arthritis: mechanisms, diagnosis and treatment
- PMID: 23007741
- PMCID: PMC4096779
- DOI: 10.1038/nrrheum.2012.153
Bone erosion in rheumatoid arthritis: mechanisms, diagnosis and treatment
Abstract
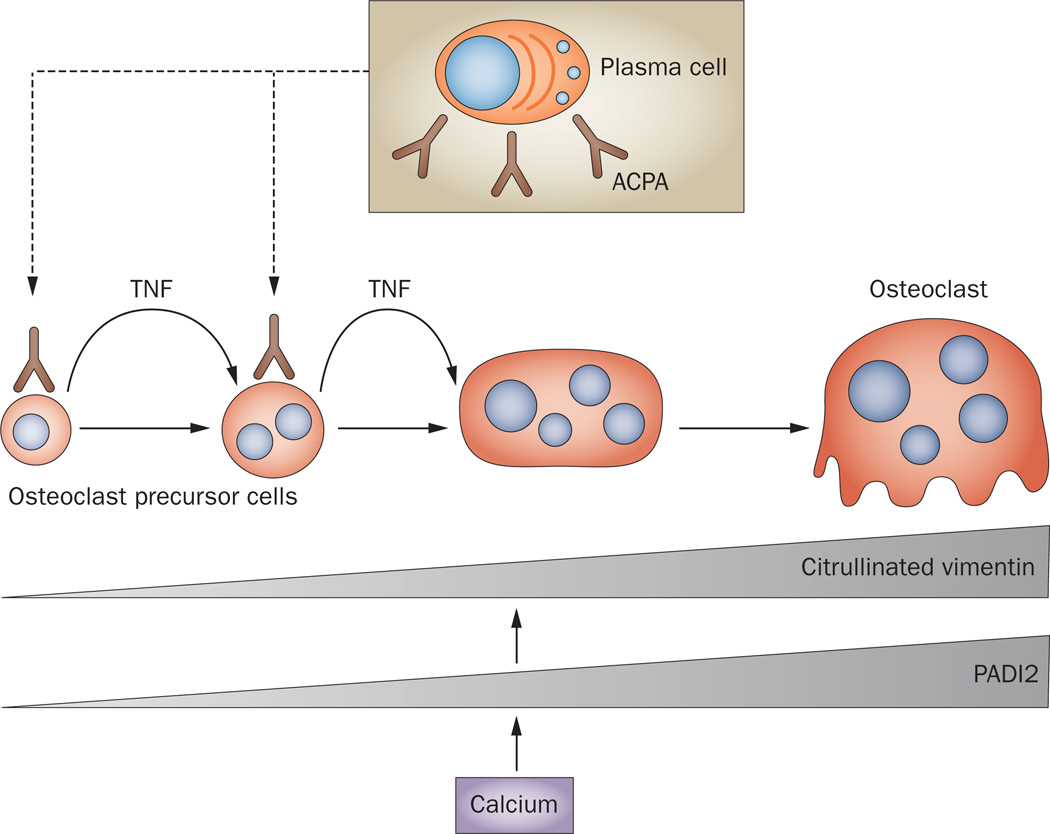
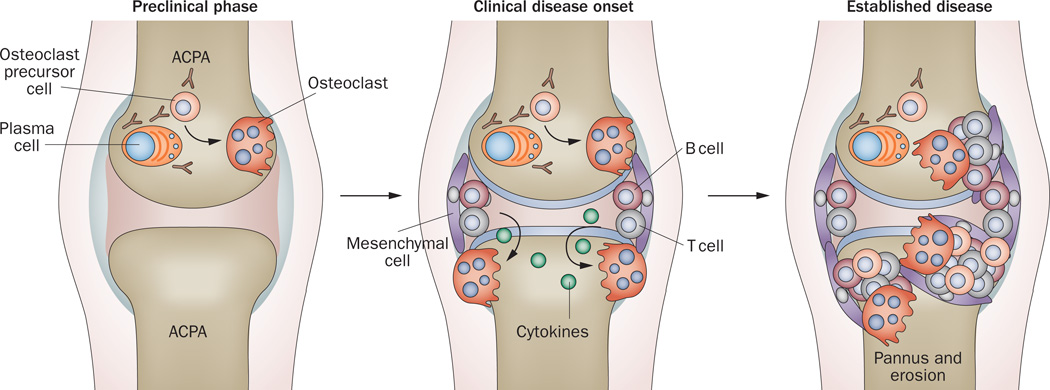
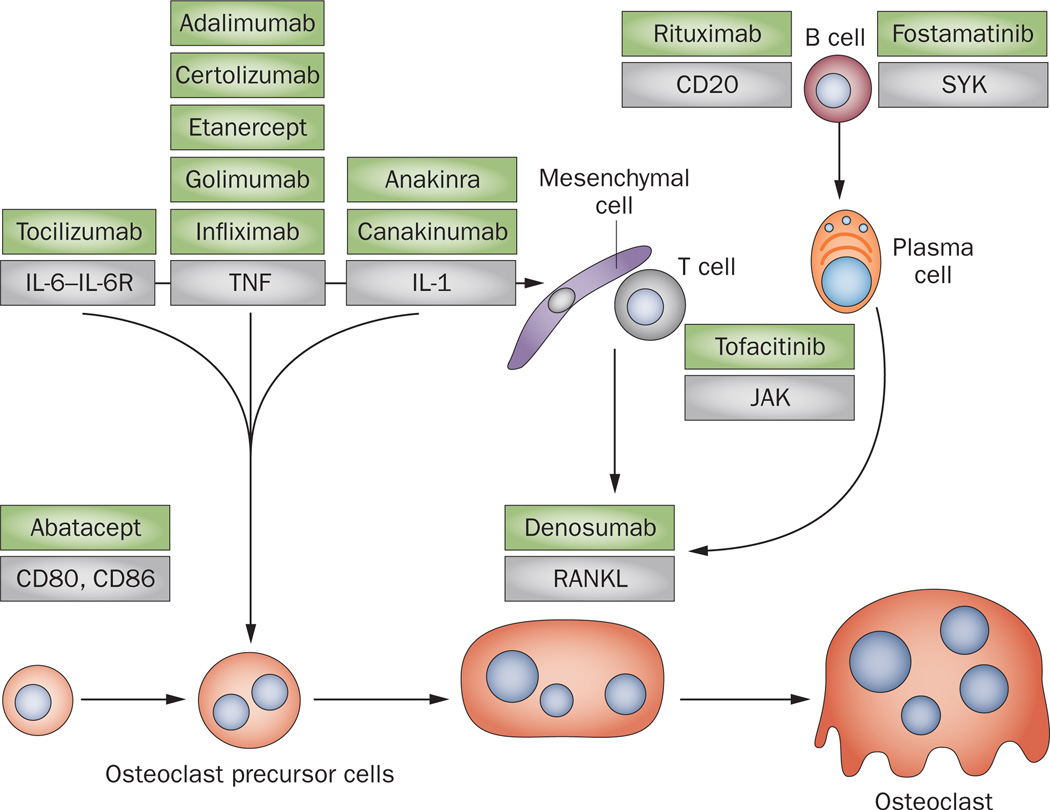
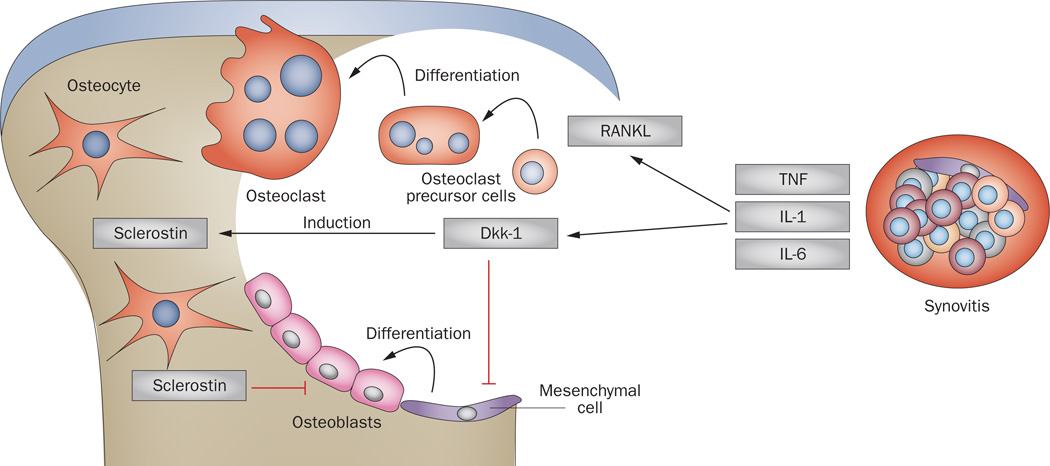
Bone erosion is a central feature of rheumatoid arthritis and is associated with disease severity and poor functional outcome. Erosion of periarticular cortical bone, the typical feature observed on plain radiographs in patients with rheumatoid arthritis, results from excessive local bone resorption and inadequate bone formation. The main triggers of articular bone erosion are synovitis, including the production of proinflammatory cytokines and receptor activator of nuclear factor κB ligand (RANKL), as well as antibodies directed against citrullinated proteins. Indeed, both cytokines and autoantibodies stimulate the differentiation of bone-resorbing osteoclasts, thereby stimulating local bone resorption. Although current antirheumatic therapy inhibits both bone erosion and inflammation, repair of existing bone lesions, albeit physiologically feasible, occurs rarely. Lack of repair is due, at least in part, to active suppression of bone formation by proinflammatory cytokines. This Review summarizes the substantial progress that has been made in understanding the pathophysiology of bone erosions and discusses the improvements in the diagnosis, monitoring and treatment of such lesions.
Figures
Similar articles
-
Rheumatoid Arthritis in the View of Osteoimmunology.Biomolecules. 2020 Dec 31;11(1):48. doi: 10.3390/biom11010048. Biomolecules. 2020. PMID: 33396412 Free PMC article. Review.
-
Pathogenesis of bone erosions in rheumatoid arthritis.Curr Opin Rheumatol. 2000 May;12(3):195-9. doi: 10.1097/00002281-200005000-00006. Curr Opin Rheumatol. 2000. PMID: 10803748 Review.
-
Fc-gamma receptors and S100A8/A9 cause bone erosion during rheumatoid arthritis. Do they act as partners in crime?Rheumatology (Oxford). 2019 Aug 1;58(8):1331-1343. doi: 10.1093/rheumatology/kez218. Rheumatology (Oxford). 2019. PMID: 31180451 Review.
-
Bone and joint destruction in rheumatoid arthritis: what is really happening?J Rheumatol Suppl. 2002 Sep;65:44-8. J Rheumatol Suppl. 2002. PMID: 12236623 Review.
-
[Inflammation and osteoclasts].Nihon Rinsho Meneki Gakkai Kaishi. 2017;40(5):367-376. doi: 10.2177/jsci.40.367. Nihon Rinsho Meneki Gakkai Kaishi. 2017. PMID: 29238019 Review. Japanese.
Cited by
-
New Horizons: Translational Aspects of Osteomorphs.J Clin Endocrinol Metab. 2024 Apr 19;109(5):e1373-e1378. doi: 10.1210/clinem/dgad711. J Clin Endocrinol Metab. 2024. PMID: 38060842 Free PMC article. Review.
-
RANK-Independent Osteoclast Formation and Bone Erosion in Inflammatory Arthritis.Arthritis Rheumatol. 2016 Dec;68(12):2889-2900. doi: 10.1002/art.39837. Arthritis Rheumatol. 2016. PMID: 27563728 Free PMC article.
-
The Contribution of Macrophage Plasticity to Inflammatory Arthritis and Their Potential as Therapeutic Targets.Cells. 2024 Sep 20;13(18):1586. doi: 10.3390/cells13181586. Cells. 2024. PMID: 39329767 Free PMC article. Review.
-
Mitochondrial Dysfunction in Rheumatoid Arthritis.Biomolecules. 2022 Sep 1;12(9):1216. doi: 10.3390/biom12091216. Biomolecules. 2022. PMID: 36139055 Free PMC article. Review.
-
The role of anti-citrullinated protein antibody in pathogenesis of RA.Clin Exp Med. 2024 Jul 8;24(1):153. doi: 10.1007/s10238-024-01359-3. Clin Exp Med. 2024. PMID: 38972923 Free PMC article. Review.
References
-
- Karsenty G, Kronenberg HM, Settembre C. Genetic control of bone formation. Annu. Rev. Cell. Dev. Biol. 2009;25:629–648. - PubMed
-
- Teitelbaum SL, Ross FP. Genetic regulation of osteoclast development and function. Nat. Rev. Genet. 2003;4:638–649. - PubMed
-
- Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423:337–342. - PubMed
-
- Baker WM. The formation of abnormal synovial cysts in the connection with the joints. St Bartolomews Hospital Reports. 1855;21:177–190.
-
- Weichselbaum A. Die feineren Veränderungen des Gelenkknorpels bei fungöser Synovitis und Karies der Gelenkenden [German] Archiv. Pathol. Anat. Physiol. Klin. Med. 1878;73:461–475.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
