

Reprogramming of DNA methylation in pollen guides epigenetic inheritance via small RNA
- PMID: 23000270
- PMCID: PMC3697483
- DOI: 10.1016/j.cell.2012.09.001
Reprogramming of DNA methylation in pollen guides epigenetic inheritance via small RNA
Abstract
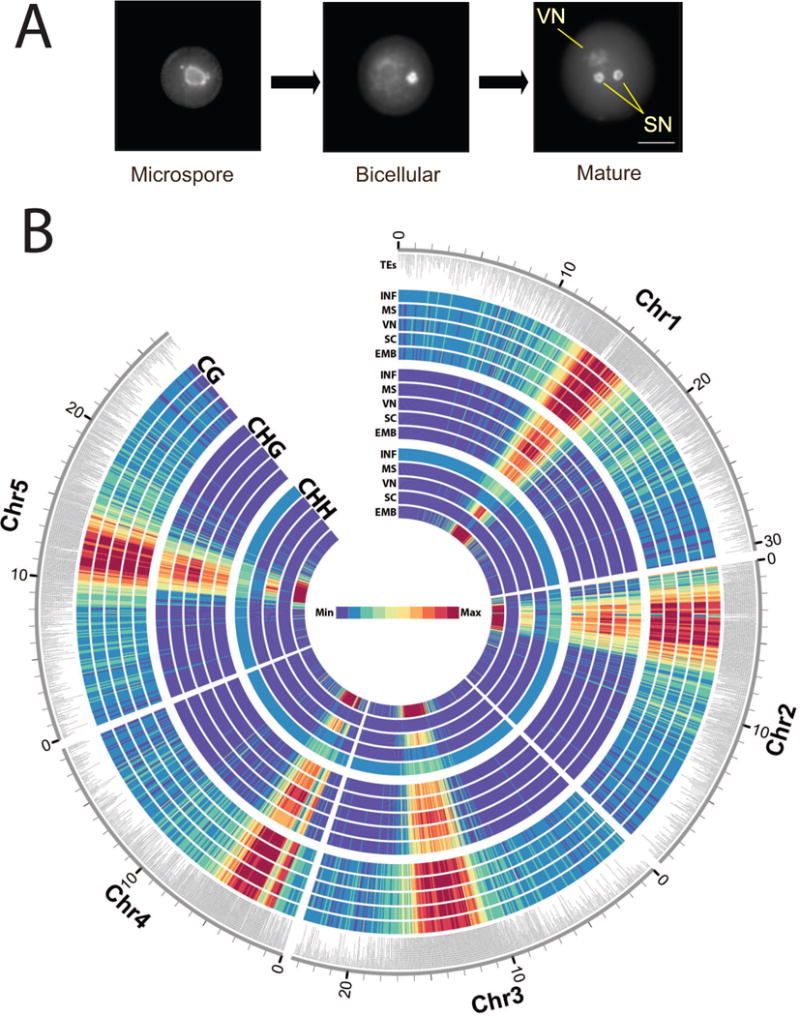
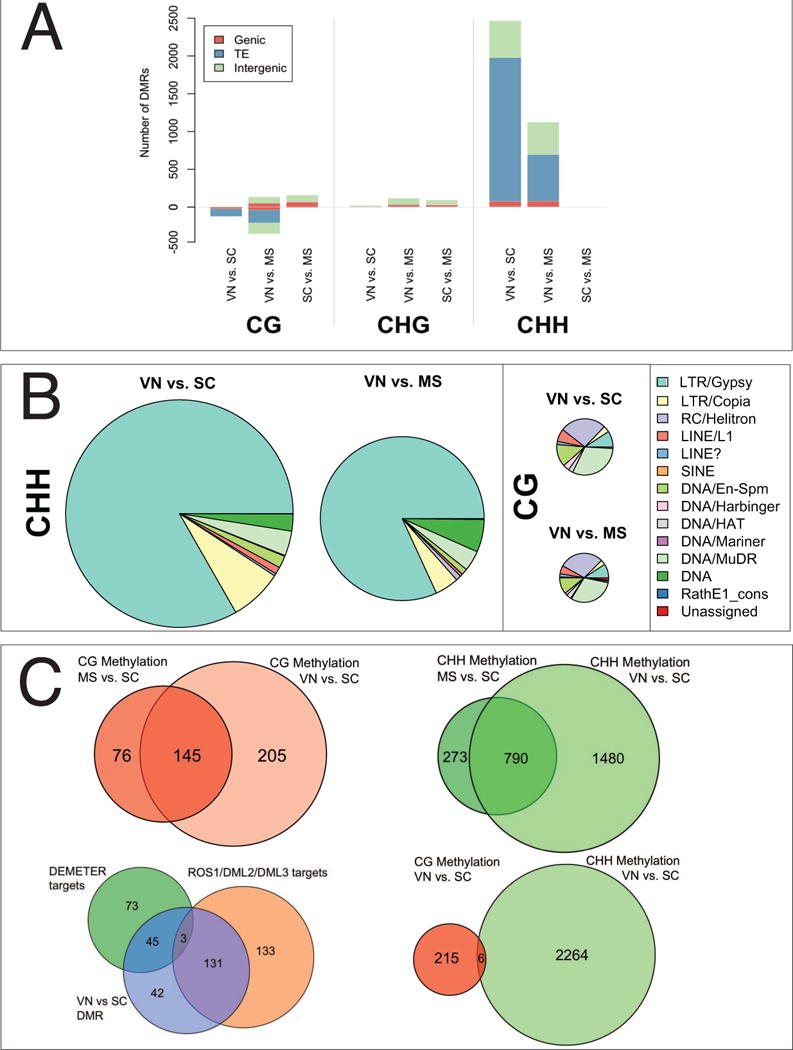
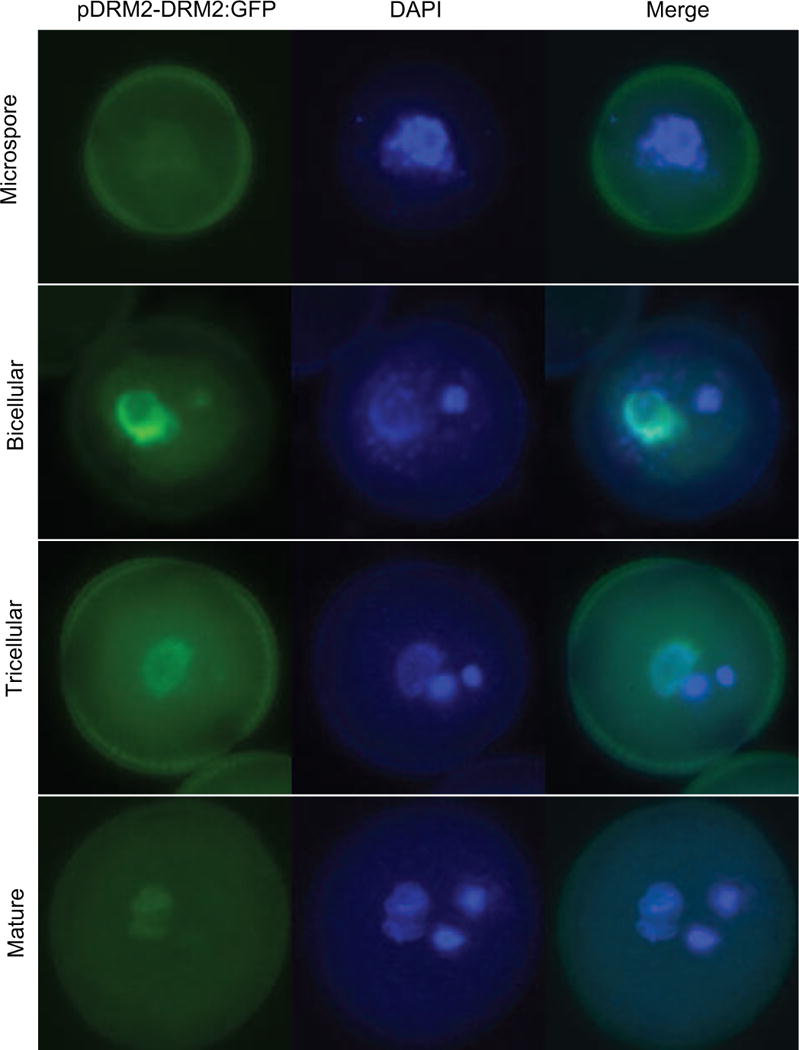
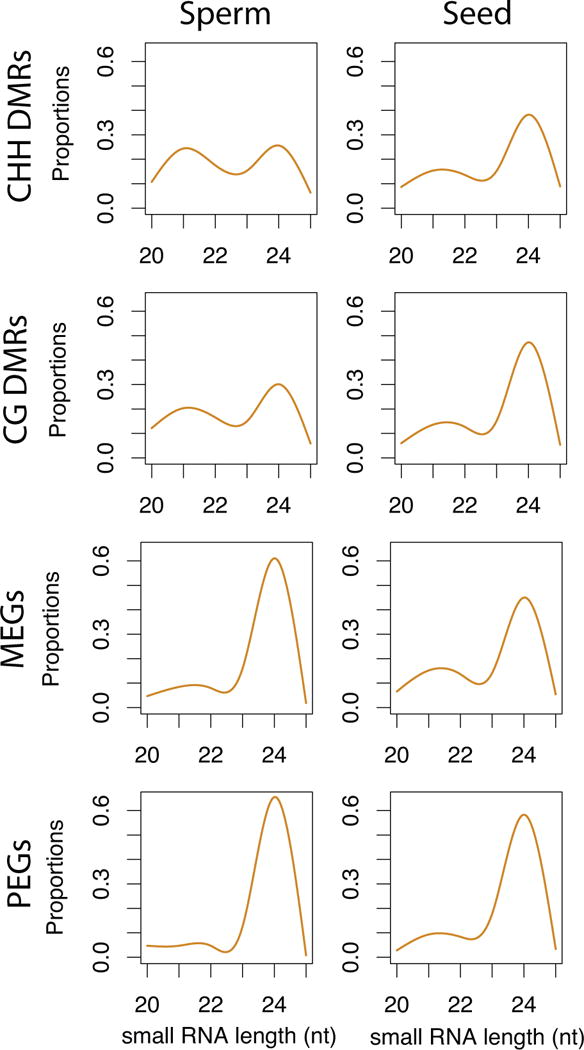
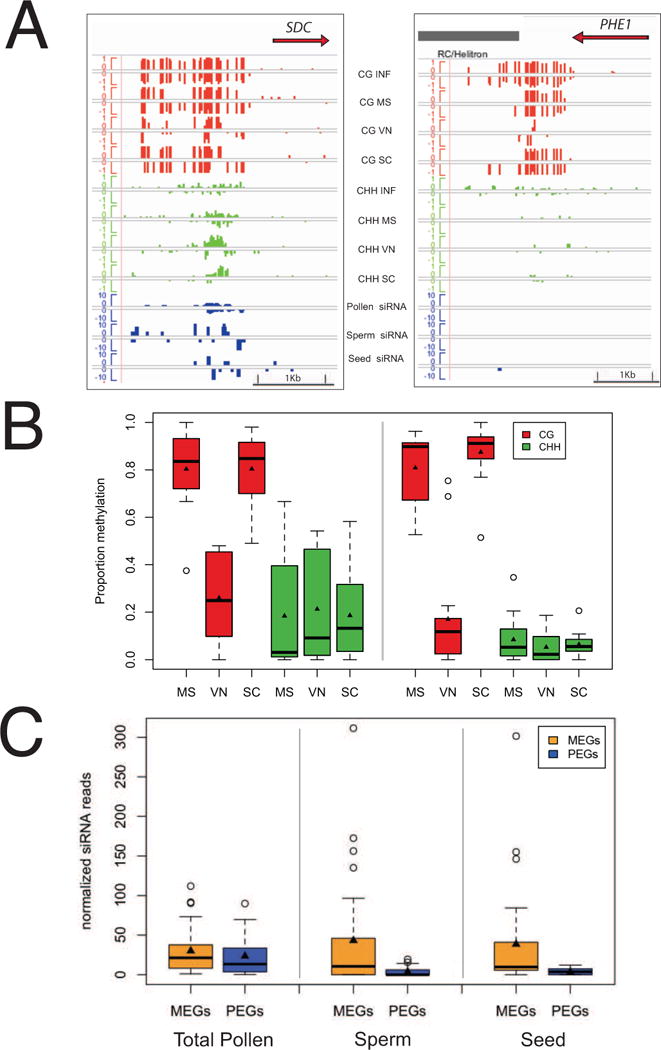
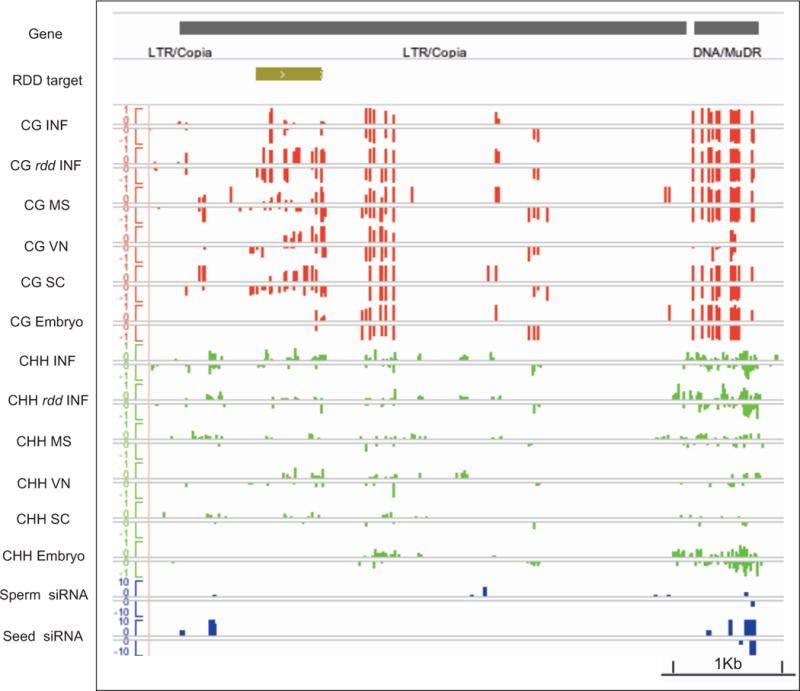
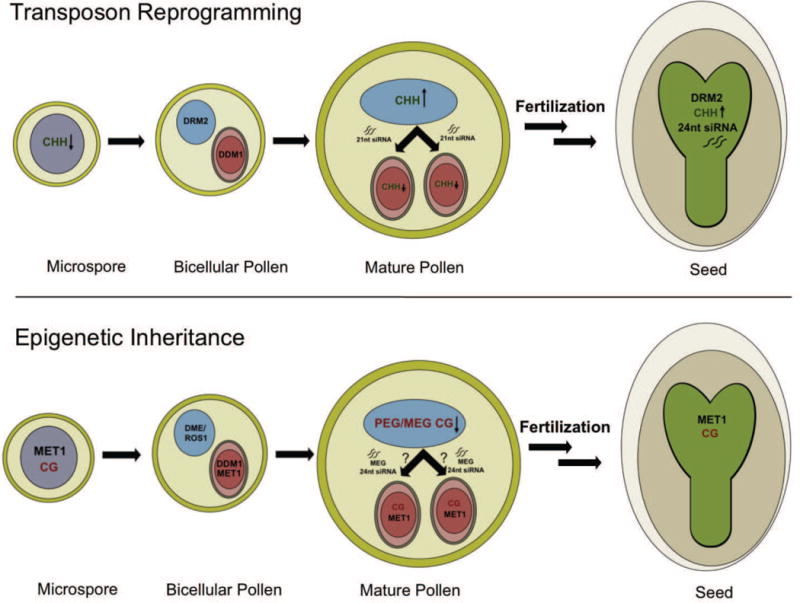
Epigenetic inheritance is more widespread in plants than in mammals, in part because mammals erase epigenetic information by germline reprogramming. We sequenced the methylome of three haploid cell types from developing pollen: the sperm cell, the vegetative cell, and their precursor, the postmeiotic microspore, and found that unlike in mammals the plant germline retains CG and CHG DNA methylation. However, CHH methylation is lost from retrotransposons in microspores and sperm cells and restored by de novo DNA methyltransferase guided by 24 nt small interfering RNA, both in the vegetative nucleus and in the embryo after fertilization. In the vegetative nucleus, CG methylation is lost from targets of DEMETER (DME), REPRESSOR OF SILENCING 1 (ROS1), and their homologs, which include imprinted loci and recurrent epialleles that accumulate corresponding small RNA and are premethylated in sperm. Thus genome reprogramming in pollen contributes to epigenetic inheritance, transposon silencing, and imprinting, guided by small RNA.
Copyright © 2012 Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Establishing epigenetic variation during genome reprogramming.RNA Biol. 2013 Apr;10(4):490-4. doi: 10.4161/rna.24085. RNA Biol. 2013. PMID: 23774895 Free PMC article.
-
Reprogramming the epigenome in Arabidopsis pollen.Cold Spring Harb Symp Quant Biol. 2012;77:1-5. doi: 10.1101/sqb.2013.77.014969. Epub 2013 Apr 25. Cold Spring Harb Symp Quant Biol. 2012. PMID: 23619015 Free PMC article. Review.
-
Epigenetic reprogramming and small RNA silencing of transposable elements in pollen.Cell. 2009 Feb 6;136(3):461-72. doi: 10.1016/j.cell.2008.12.038. Cell. 2009. PMID: 19203581 Free PMC article.
-
The maize methylome influences mRNA splice sites and reveals widespread paramutation-like switches guided by small RNA.Genome Res. 2013 Oct;23(10):1651-62. doi: 10.1101/gr.153510.112. Epub 2013 Jun 5. Genome Res. 2013. PMID: 23739895 Free PMC article.
-
siRNAs and DNA methylation: seedy epigenetics.Trends Plant Sci. 2010 Apr;15(4):204-10. doi: 10.1016/j.tplants.2010.01.002. Epub 2010 Feb 1. Trends Plant Sci. 2010. PMID: 20129810 Review.
Cited by
-
Predictable and stable epimutations induced during clonal plant propagation with embryonic transcription factor.PLoS Genet. 2022 Nov 16;18(11):e1010479. doi: 10.1371/journal.pgen.1010479. eCollection 2022 Nov. PLoS Genet. 2022. PMID: 36383565 Free PMC article.
-
An AP endonuclease functions in active DNA demethylation and gene imprinting in Arabidopsis [corrected].PLoS Genet. 2015 Jan 8;11(1):e1004905. doi: 10.1371/journal.pgen.1004905. eCollection 2015 Jan. PLoS Genet. 2015. PMID: 25569774 Free PMC article.
-
Prodigious plant methylomes.Genome Biol. 2016 Sep 27;17(1):197. doi: 10.1186/s13059-016-1065-2. Genome Biol. 2016. PMID: 27677311 Free PMC article.
-
Chromatin regulates expression of small RNAs to help maintain transposon methylome homeostasis in Arabidopsis.Genome Biol. 2020 Sep 17;21(1):251. doi: 10.1186/s13059-020-02163-4. Genome Biol. 2020. PMID: 32943088 Free PMC article.
-
Gametophytic epigenetic regulators, MEDEA and DEMETER, synergistically suppress ectopic shoot formation in Arabidopsis.Plant Cell Rep. 2024 Feb 11;43(3):68. doi: 10.1007/s00299-024-03159-1. Plant Cell Rep. 2024. PMID: 38341844
References
-
- Becker C, Hagmann J, Muller J, Koenig D, Stegle O, Borgwardt K, Weigel D. Spontaneous epigenetic variation in the Arabidopsis thaliana methylome. Nature. 2011;480:245–249. - PubMed
-
- Berger F, Twell D. Germline specification and function in plants. Annu Rev. Plant Biol. 2011;62:461–484. - PubMed
-
- Boavida LC, Becker JD, Feijo JA. The making of gametes in higher plants. Int J Dev Biol. 2005;49:595–614. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
