

Review
doi: 10.1038/nrm3434.
Epub 2012 Sep 20.
TGFβ signalling in context
Affiliations
- PMID: 22992590
- PMCID: PMC4027049
- DOI: 10.1038/nrm3434
Item in Clipboard
Review
TGFβ signalling in context
Nat Rev Mol Cell Biol.
2012 Oct.
Abstract
The basic elements of the transforming growth factor-β (TGFβ) pathway were revealed more than a decade ago. Since then, the concept of how the TGFβ signal travels from the membrane to the nucleus has been enriched with additional findings, and its multifunctional nature and medical relevance have relentlessly come to light. However, an old mystery has endured: how does the context determine the cellular response to TGFβ? Solving this question is key to understanding TGFβ biology and its many malfunctions. Recent progress is pointing at answers.
Figures
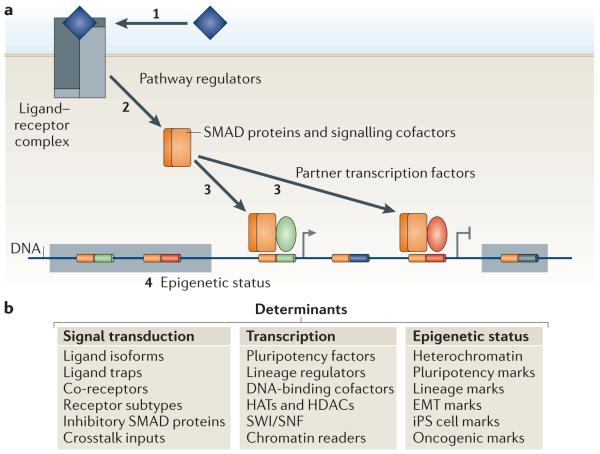
a | Three types of contextual determinants shape the transforming growth factor-β (TGFβ)-mediated transcriptional response in a cell. First, a large number of signal transduction regulatory factors determine the access of TGFβ ligands to signalling receptors (1), and of receptors to SMAD proteins and other signal delivery factors (2). Second, transcription factors, histone readers and modifiers and chromatin remodellers that bind to activated SMAD proteins determine what genes will be targeted by the signal transduction complexes and whether expression of the target genes will be positively or negatively regulated (3). The third type of contextual determinants is presented by the epigenetic status of the cell. The epigenetic state dictates whether genes are in an active `open' chromatin conformation (and are therefore accessible for SMAD complexes), in a repressive chromatin conformation (and therefore in a silenced `closed' state that is not accessible for transcriptional regulation), or whether these genes are in a poised chromatin state that is silenced yet responsive to TGFβ signalling and the appropriate chromatin readers (4). Composite enhancer elements are represented as bi-coloured segments on the DNA, inside a shaded box if the genes are in repressive chromatin, and unoccupied if the cell does not express the required DNA-binding SMAD cofactor. b | A list of the contextual determinants that affect the signal transduction and transcription steps and the regulators of the epigenetic status. EMT, epithelial–mesenchymal transition; HATs, histone acetyl transferases; HDACs, histone deacetylases; iPS cell, induced pluripotent stem cell; SWI/SNF, Switch/sucrose nonfermentable.
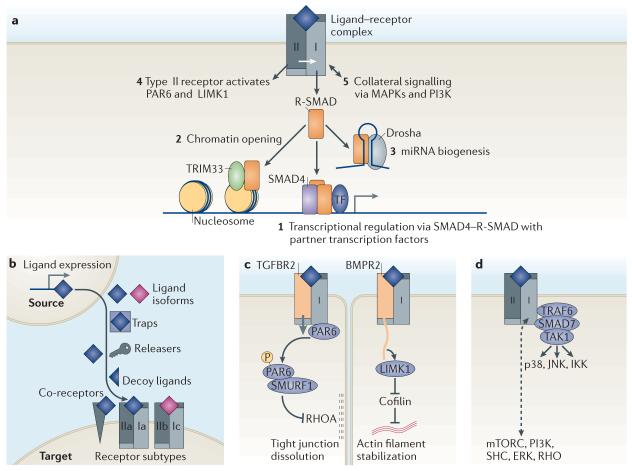
a | Transforming growth factor-β (TGFβ) family ligands signal by assembling a hetero-tetrameric receptor complex with two type I (which are the main signal propagators) receptor components (shown in light grey) and two type II (which are activators) components (shown in dark grey). Signalling is mediated via a cytoplasmic Ser/Thr kinase domain. Signalling modality 1 is the canonical SMAD pathway, and modality 2 its companion. Signalling modalities 3–5 are considered as non-canonical TGFβ signalling pathways. Receptor-phosphorylated SMAD proteins (R-SMAD proteins) form transcriptional complexes that pair with other context-dependent transcription factors to regulate hundreds of genes (1) (for details, see FIGS 3,5). Activated R-SMAD proteins can also form a complex with TRIM33 (tripartite motif containing 33) that recognizes certain histone marks and disables their repressive action, which results in chromatin opening and thereby allows access for canonical SMAD complexes (2) (for details, see FIG. 4). R-SMAD proteins can participate in microRNA (miRNA) processing by Drosha complexes for the biogenesis of a subset of SMAD-binding miRNA precursors (3). TGFβ and bone morphogenetic protein (BMP) type II receptors signal by directly activating partitioning defective 6 (PAR6) and LIM kinase 1 (LIMK1), respectively (4). TGFβ and BMP receptors can activate various mitogen-activated protein kinase (MAPKs) and phosphoinositide 3-kinase (PI3K) pathways (5). b | Seven classes of determinants regulate the access of ligand to TGFβ receptors. These include: the level of expression of a ligand in source cells, the type of ligand isoforms that is available, factors that can sequester the ligands (termed traps) or release them (termed releasers), decoy ligands that occupy the receptors or co-receptors without triggering signalling and the type of receptor and co-receptor that is expressed in target cells. c | Direct signalling by type II receptor kinases. TGFβ receptor type II (TGFBR2) phosphorylates the tight junction regulator PAR6 to recruit the E3 ubiquitin ligase SMURF1 and target RHOA for degradation. This leads to dissolution of tight junctions in epithelial cells. BMP receptor type II (BMPR2) contains a long carboxy-terminal tail that binds and activates LIMK1, thereby inhibiting the actin-disassembling protein cofilin. This results in stabilization of actin filaments. d | TRAF6 (tumour necrosis factor receptor-associated factor 6) acts together with SMAD7 and both are known binding partners for TGFβ receptors. The interaction between TRAF6 and SMAD7 is implicated in the activation of TAK1 (TGFβ-activated kinase 1), which is a protein kinase upstream of the signal transduction kinases p38, JNK (Jun amino-terminal kinase) and IKK (inhibitor of κB kinase). TGFβ and BMP receptors can activate several other signal transducers (such as mTORC (mammalian target of rapamycin), PI3K, SHC (SH2 domain-containing transforming protein), ERK (extracellular signal-regulated kinase) and RHO), although the biochemical and structural bases for many of these links remain unknown.
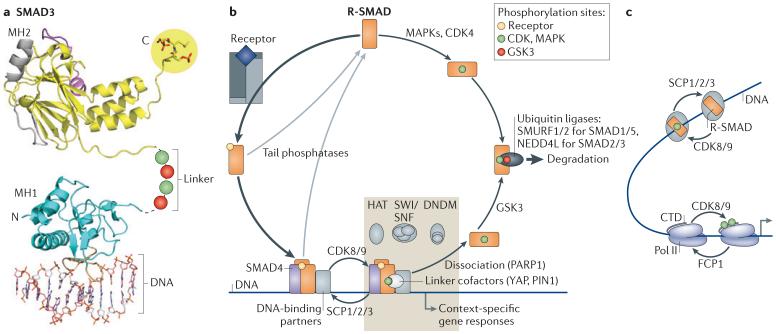
a | Structure of SMAD3 as a representative of receptor-phosphorylated SMAD proteins (R-SMAD proteins). SMAD proteins consist of two globular domains (termed MH1 and MH2) that are coupled by a linker region. The signalling receptors phosphorylate R-SMAD proteins at the carboxy-terminal sequence Ser-X-Ser (where X can be any amino acid), creating an acidic tail that allows binding to SMAD4 (not shown). The linker is phosphorylated by cyclin-dependent kinases (CDKs), mitogen-activated protein kinases (MAPKs) (green) and glycogen synthase kinase 3 (GSK3) (red). The phosphorylated linker creates a docking sites for positive and negative regulators of SMAD function. b | Following phosphorylation by receptors (yellow), R-SMAD proteins bind SMAD4 and form a hetero-trimeric complex that binds to DNA with partner transcription factors. The transcriptional kinases CDK8 and CDK9 phosphorylate the linker region for peak activation (green). Key participants in the SMAD transcriptional complex include the SWI/SNF nucleosome positioning complex, the HATs (histone acetyl transferases) p300 and CBP (cyclic AMP response element-binding protein) and in certain target genes a DNA demethylating complex (DNDM). Repressive SMAD complexes recruit histone deacetylases (HDACs) through co-repressors (not shown). Various linker-bound factors also participate. Small C-terminal domain (CTD) phosphatases (SCPs) dephosphorylate the linker, allowing repeated utilization of the activated SMAD complex. Poly(ADP-ribose) polymerase 1 (PARP1) and other factors mediate the dissociation of this complex, and tail phosphatases return R-SMAD proteins to the basal state. If R-SMAD proteins are not dephosphorylated, GSK3 recognizes R-SMAD proteins phosphorylated by CDK8 and CDK9 and further increases R-SMAD phosphorylation (red), thereby marking R-SMAD proteins for recognition by E3 ubiquitin ligases and proteasome-mediated degradation. In this manner, SMAD transcriptional action becomes coupled to SMAD turnover. Mitogens and stresses acting through MAPKs, and the cell cycle acting through CDK4, can also phosphorylate the linker to limit the availability of R-SMAD proteins for TGFβ or bone morphogenetic protein (BMP) signalling. c | Intriguing parallels exist between the SMAD andRNA polymerase II (Pol II) transcriptional cycles. R-SMAD proteins and Pol II are phosphorylated (green) by the same kinases (which are CDK8 and CDK9) for peak activation and dephosphorylated by structurally related phosphatases (SCP1, SCP2 and SCP3) that reset the basal state. FCP1, transcription factor IIF-associated CTD phosphatase 1; NEDD4L, neural precursor cell expressed developmentally downregulated protein 4-like; PIN1, peptidylprolyl cis/trans isomerase, NIMA-interacting 1; SMURF, SMAD-specific E3 ubiquitin protein ligase 1; YAP, Yes-associated protein. Image in part a is reproduced, with permission, from REF. 63 © (2011) Cold Spring Harbor Laboratory Press.
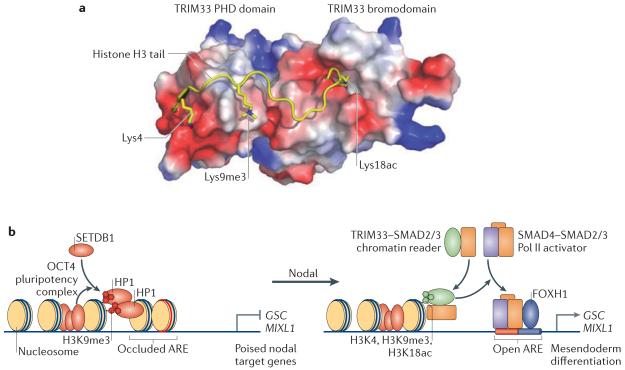
a | TRIM33 (tripartite motif containing 33) is a chromatin reader that recognizes a triple feature on histone H3: unmodified Lys4, trimethylated Lys9 (Lys9me3) and acetylated Lys18 (Lys18ac), as seen in the crystal structure of the TRIM33 plant homeodomain PHD-bromo cassette bound to the cognate histone peptide. b | The OCT4 pluripotency complex (which contains OCT4, SOX2 and NANOG) in embryonic stem cells prompts SETDB1 (SET-domain binding 1) to trimethylate histone H3 at Lys9 (H3K9me3) at the promoters of the mesendoderm differentiation genes goosecoid homeobox (GSC), mix paired-like homeobox (MIXL1) and other genes. H3K9me3 recruits the chromatin compacting factor heterochromatin protein 1 (HP1) to implement repression. Under differentiation conditions, GSC and MIXL1 are silent but poised for activation by Nodal signals. The gene promoters present cognate histone marks that are recognized by TRIM33. Nodal drives the formation of two companion complexes, TRIM33–SMAD2/3 and SMAD4–SMAD2/3. The TRIM33–SMAD2/3 complex binds to the poised nucleosomes, displacing HP1, thus enabling SMAD4–SMAD2/3 and their partner in this context, FOXH1 (forkhead box H1), to co-occupy cognate DNA elements (termed activin response elements (AREs)) and activate transcription. Image in part a is courtesy of Z. Wang and D. J. Patel, Memorial Sloan-Kettering Cancer Center, New York, New York, USA.
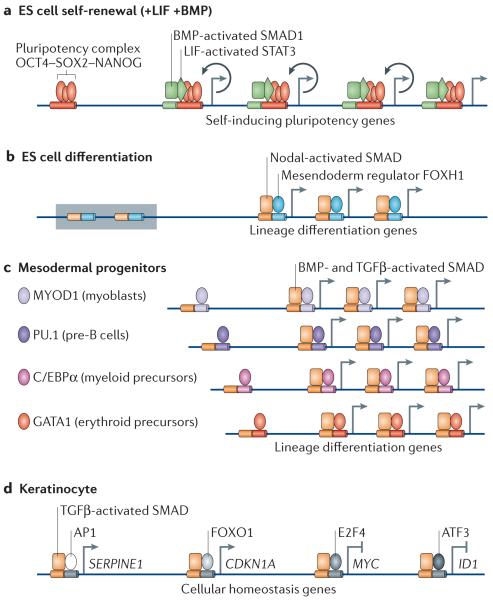
a | In embryonic stem (ES) cells, OCT4, SOX2 and NANOG form the core of a self-renewal network that is stimulated by the bone morphogenetic protein (BMP) mediator SMAD1 and the leukaemia inhibitory factor (LIF) mediator STAT3. SMAD1, STAT3 and the OCT4 complex co-occupy many active sites throughout the genome, including OCT4, SOX2 and NANOG themselves, to enforce self-renewal (indicated by circular arrows). b | In ES cells that lack self-renewal signals, the Nodal-activated SMAD4–SMAD2/3 complex, together with the TRIM33 (tripartite motif containing 33)–SMAD2/3 complex (see FIG. 4), activates mesendoderm differentiation genes in poised chromatin (indicated by a shaded box). Forkhead box H1 (FOXH1) is a mesendoderm lineage factor that recruits the SMAD4 complex to multiple differentiation genes. c | In lineage-restricted progenitors, lineage identity factors are dominant partners of transforming growth factor-β (TGFβ)- or BMP-activated SMAD4 complexes and co-occupy the genome to implement differentiation. d | In differentiated cells, TGFβ activated SMAD4–SMAD2/3 complexes are recruited by different partner transcription factors to different subsets of target genes (each subset is represented in the figure by only one gene). The combination of these pathways that lead to transcriptional regulation constitutes the overall TGFβ-mediated transcriptional response for regulation of cell proliferation, adhesion, extracellular matrix properties, the secretome and other cell homeostasis functions in any cell type. The diagram shows the factors that regulate homeostasis of a keratinocyte (that is, a differentiated ectodermal derivative). ATF3, activating transcription factor 3; AP1, adaptor protein 1; C/EBPα, CCAAT/enhancer-binding protein-α; CDKN1A, cyclin-dependent kinase inhibitor 1A; ID1, inhibitor of DNA binding 1; MYOD1, myoblast determination protein 1.
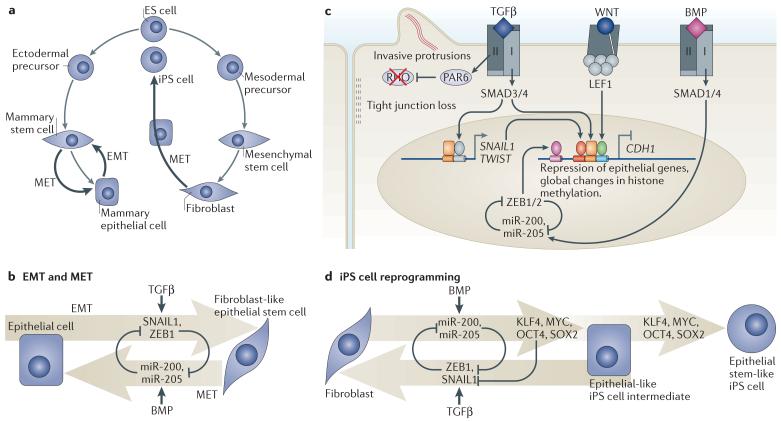
a | Cell reprogramming processes regulated by transforming growth factor-β (TGFβ) and bone morphogenetic protein (BMP) include epithelial–mesenchymal transition (EMT) in mammary epithelial cells, which can generate epithelial stem-like cells, and the reverse process, mesenchymal–epithelial transition (MET). Reprogramming of fibroblasts into induced pluripotent stem (iPS) cells involves an intermediate MET-like process that is also regulated by BMP and TGFβ. b | EMT is driven by the core regulators SNAIL1, SNAIL2, ZEB1 (zinc-finger E-box binding factor 1) and ZEB2 (SNAIL2 and ZEB2 are not shown for simplicity) and other factors. EMT is inhibited by the pro-epithelial microRNAs miR-200 and miR-205, which in turn are suppressed by ZEB1. In mammary epithelial cells, EMT is stimulated by TGFβ and opposed by BMP. c | WNT primes epithelial cells to undergo EMT in response to TGFβ. TGFβ triggers SMAD-dependent induction of SNAIL1 and TWIST. SMAD3 and SMAD4 then join SNAIL1 and WNT-activated LEF1 (lymphoid enhancer-binding factor 1) to co-occupy the promoter of CDH1 (which is the gene encoding epithelial cadherin (E-cadherin)) for repression of this key epithelial gene. This and an associated genome-wide chromatin changes implement the mesenchymal phenotype. In parallel, TGFβ receptor type II (TGFBR2) phosphorylates partitioning defective 6 (PAR6) to mediate dissolution of tight junctions and foster migration (see FIG. 2c). BMP signalling through SMAD1 and SMAD4 upregulates miR-200 and miR-205 to oppose EMT. d | Fibroblast reprogramming into iPS cells by ectopic expression of OCT4, Krueppel-like factor 4 (KLF4), SOX2 and MYC requires MET, which is facilitated by BMP-induced miR-200 and miR-205 expression. By interfering with MET, TGFβ suppresses the generation of iPS cells. The use of TGFBR1 inhibitors increases the efficiency of iPS cell generation and reduces the requirement for MYC and SOX2.
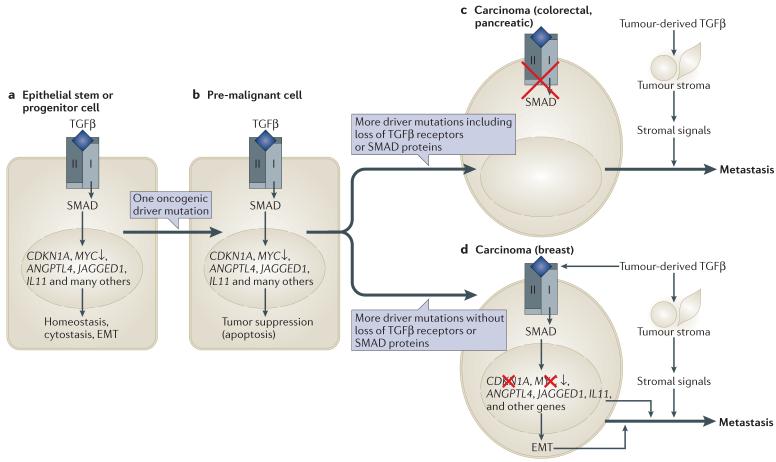
a | Transforming growth factor-β (TGFβ)-activated SMAD proteins regulate hundreds of genes in normal epithelial cells, including cytostatic genes and genes involved in homeostasis. The examples include genes that are upregulated by TGFβ, except MYC, which is downregulated. b | When a stem or progenitor cell incurs an oncogenic mutation (for example, loss of APC (adenomatous polyposis coli) tumour suppressor, activation of KRAS or amplification of HER2) it becomes liable to undergo apoptosis if exposed to TGFβ. To advance in the tumorigenic path, this cell must accumulate additional alterations that, among other achievements, disable the tumour suppressive responsiveness of the cell to TGFβ. c | One path to tumour formation involves the selection of malignant clones that have lost TGFβ signalling owing to mutations in TGFBR1 (TGF beta receptor type II), TGFBR2 or SMAD4. This outcome is frequent in colorectal and pancreatic carcinomas. As a result, cancer cells can withstand a TGFβ-rich tumour stroma and benefit from pro-tumorigenic effects such as TGFβ induction of stroma-derived cytokines that promote cell survival. d | Another path involves the selection of clones that have lost tumour suppressive TGFβ responses but retain an intact SMAD signalling machinery. This outcome is frequent in breast carcinomas, gliomas and melanomas. As a result, cancer cells not only withstand a TGFβ-rich microenvironment but also respond to TGFβ, resulting in SMAD-dependent gene responses that in this context are profitable for metastasis. Examples include the induction of angiopoietin-like 4 (ANGPTL4) that primes breast cancer cells for extravasation, increased expression of interleukin 11 (IL11) and NOTCH ligand JAGGED1 that allows cancer cells in the bone marrow to activate osteoclasts for osteolytic metastasis, and epithelial–mesenchymal transition (EMT) that provides an invasive and tumour initiating phenotype.
Similar articles
-
USP11 augments TGFβ signalling by deubiquitylating ALK5.Open Biol. 2012 Jun;2(6):120063. doi: 10.1098/rsob.120063. Open Biol. 2012. PMID: 22773947 Free PMC article.
-
Nitric oxide induces TIMP-1 expression by activating the transforming growth factor beta-Smad signaling pathway.J Biol Chem. 2005 Nov 25;280(47):39403-16. doi: 10.1074/jbc.M504140200. Epub 2005 Sep 23. J Biol Chem. 2005. PMID: 16183640
-
TGFβ receptor endocytosis and Smad signaling require synaptojanin1, PI3K-C2α-, and INPP4B-mediated phosphoinositide conversions.Mol Biol Cell. 2020 Mar 1;31(5):360-372. doi: 10.1091/mbc.E19-11-0662. Epub 2020 Jan 8. Mol Biol Cell. 2020. PMID: 31913757 Free PMC article.
-
Regulated intramembrane proteolysis of the TGFβ type I receptor conveys oncogenic signals.Future Oncol. 2014 Aug;10(11):1853-61. doi: 10.2217/fon.14.45. Epub 2014 Mar 5. Future Oncol. 2014. PMID: 24597658 Review.
-
Regulating the stability of TGFbeta receptors and Smads.Cell Res. 2009 Jan;19(1):21-35. doi: 10.1038/cr.2008.308. Cell Res. 2009. PMID: 19030025 Review.
Cited by
-
Doxorubicin restrains osteogenesis and promotes osteoclastogenesis in vitro.Am J Transl Res. 2020 Sep 15;12(9):5640-5654. eCollection 2020. Am J Transl Res. 2020. PMID: 33042445 Free PMC article.
-
Characterization of the roles of amphiregulin and transforming growth factor β1 in microvasculature-like formation in human granulosa-lutein cells.Front Cell Dev Biol. 2022 Aug 24;10:968166. doi: 10.3389/fcell.2022.968166. eCollection 2022. Front Cell Dev Biol. 2022. PMID: 36092732 Free PMC article.
-
Bacterial Lipopolysaccharide Induces PD-L1 Expression and an Invasive Phenotype of Oral Squamous Cell Carcinoma Cells.Cancers (Basel). 2024 Jan 13;16(2):343. doi: 10.3390/cancers16020343. Cancers (Basel). 2024. PMID: 38254832 Free PMC article.
-
Adamts10 controls transforming growth factor β family signaling that contributes to retinal ganglion cell development.Front Mol Biosci. 2022 Sep 6;9:989851. doi: 10.3389/fmolb.2022.989851. eCollection 2022. Front Mol Biosci. 2022. PMID: 36148008 Free PMC article.
-
Direct Regulation of Alternative Splicing by SMAD3 through PCBP1 Is Essential to the Tumor-Promoting Role of TGF-β.Mol Cell. 2016 Nov 3;64(3):549-564. doi: 10.1016/j.molcel.2016.09.013. Epub 2016 Oct 13. Mol Cell. 2016. PMID: 27746021 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
