

De novo assembly of highly diverse viral populations
- PMID: 22974120
- PMCID: PMC3469330
- DOI: 10.1186/1471-2164-13-475
De novo assembly of highly diverse viral populations
Abstract
Background: Extensive genetic diversity in viral populations within infected hosts and the divergence of variants from existing reference genomes impede the analysis of deep viral sequencing data. A de novo population consensus assembly is valuable both as a single linear representation of the population and as a backbone on which intra-host variants can be accurately mapped. The availability of consensus assemblies and robustly mapped variants are crucial to the genetic study of viral disease progression, transmission dynamics, and viral evolution. Existing de novo assembly techniques fail to robustly assemble ultra-deep sequence data from genetically heterogeneous populations such as viruses into full-length genomes due to the presence of extensive genetic variability, contaminants, and variable sequence coverage.
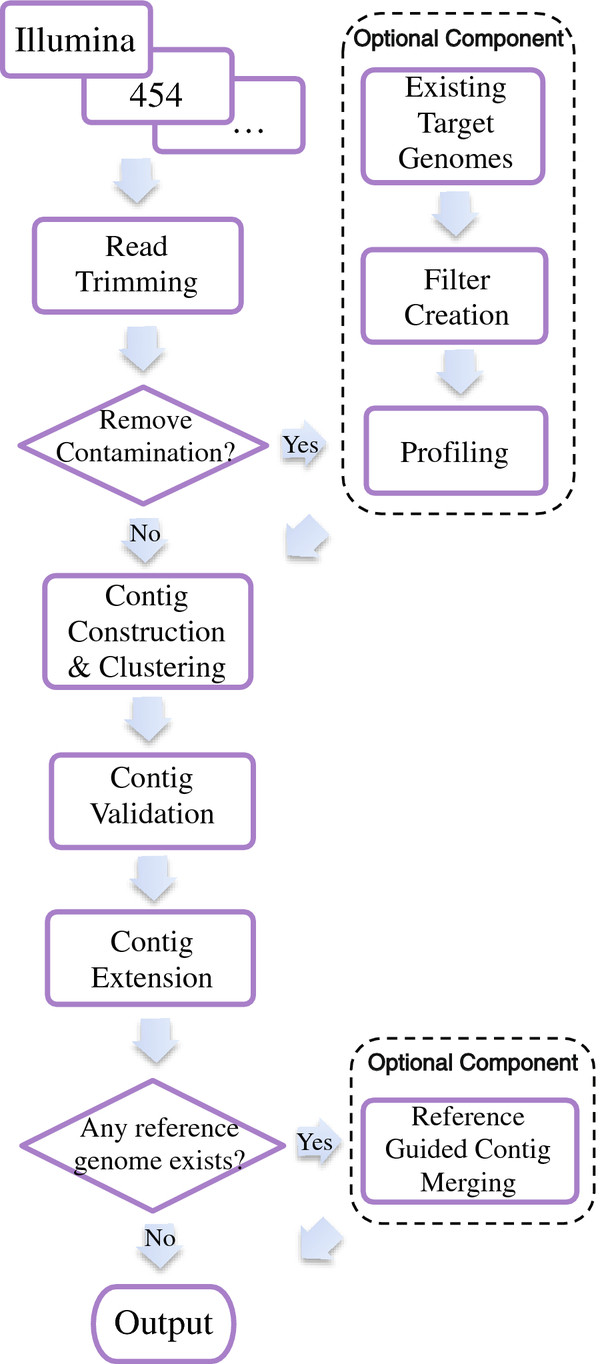
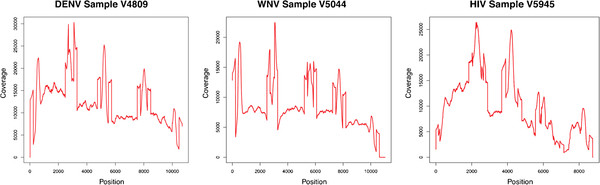
Results: We present VICUNA, a de novo assembly algorithm suitable for generating consensus assemblies from genetically heterogeneous populations. We demonstrate its effectiveness on Dengue, Human Immunodeficiency and West Nile viral populations, representing a range of intra-host diversity. Compared to state-of-the-art assemblers designed for haploid or diploid systems, VICUNA recovers full-length consensus and captures insertion/deletion polymorphisms in diverse samples. Final assemblies maintain a high base calling accuracy. VICUNA program is publicly available at: http://www.broadinstitute.org/scientific-community/science/projects/viral-genomics/ viral-genomics-analysis-software.
Conclusions: We developed VICUNA, a publicly available software tool, that enables consensus assembly of ultra-deep sequence derived from diverse viral populations. While VICUNA was developed for the analysis of viral populations, its application to other heterogeneous sequence data sets such as metagenomic or tumor cell population samples may prove beneficial in these fields of research.
Figures
Similar articles
-
V-Phaser 2: variant inference for viral populations.BMC Genomics. 2013 Oct 3;14:674. doi: 10.1186/1471-2164-14-674. BMC Genomics. 2013. PMID: 24088188 Free PMC article.
-
Viral deep sequencing needs an adaptive approach: IRMA, the iterative refinement meta-assembler.BMC Genomics. 2016 Sep 5;17(1):708. doi: 10.1186/s12864-016-3030-6. BMC Genomics. 2016. PMID: 27595578 Free PMC article.
-
Benchmarking and Assessment of Eight De Novo Genome Assemblers on Viral Next-Generation Sequencing Data, Including the SARS-CoV-2.OMICS. 2022 Jul;26(7):372-381. doi: 10.1089/omi.2022.0042. Epub 2022 Jun 28. OMICS. 2022. PMID: 35759429
-
Bioinformatics tools for analysing viral genomic data.Rev Sci Tech. 2016 Apr;35(1):271-85. doi: 10.20506/rst.35.1.2432. Rev Sci Tech. 2016. PMID: 27217183 Review.
-
The present and future of de novo whole-genome assembly.Brief Bioinform. 2018 Jan 1;19(1):23-40. doi: 10.1093/bib/bbw096. Brief Bioinform. 2018. PMID: 27742661 Review.
Cited by
-
IAVCP (Influenza A Virus Consensus and Phylogeny): Automatic Identification of the Genomic Sequence of the Influenza A Virus from High-Throughput Sequencing Data.Viruses. 2024 May 29;16(6):873. doi: 10.3390/v16060873. Viruses. 2024. PMID: 38932165 Free PMC article.
-
A novel computational pipeline for var gene expression augments the discovery of changes in the Plasmodium falciparum transcriptome during transition from in vivo to short-term in vitro culture.Elife. 2024 Jan 25;12:RP87726. doi: 10.7554/eLife.87726. Elife. 2024. PMID: 38270586 Free PMC article.
-
Validation of Variant Assembly Using HAPHPIPE with Next-Generation Sequence Data from Viruses.Viruses. 2020 Jul 14;12(7):758. doi: 10.3390/v12070758. Viruses. 2020. PMID: 32674515 Free PMC article.
-
A method for near full-length amplification and sequencing for six hepatitis C virus genotypes.BMC Genomics. 2016 Mar 17;17:247. doi: 10.1186/s12864-016-2575-8. BMC Genomics. 2016. PMID: 26988550 Free PMC article.
-
High resolution sequencing of hepatitis C virus reveals limited intra-hepatic compartmentalization in end-stage liver disease.J Hepatol. 2017 Jan;66(1):28-38. doi: 10.1016/j.jhep.2016.07.048. Epub 2016 Aug 13. J Hepatol. 2017. PMID: 27531641 Free PMC article.
References
-
- Henn MR, Boutwell CL, Charlebois P, Lennon NJ, Power KA, Macalalad AR, Berlin AM, Malboeuf CM, Ryan EM, Gnerre S, Zody MC, Erlich RL, Green LM, Berical A, Wang Y, Casali M, Streeck H, Bloom AK, Dudek T, Tully D, Newman R, Axten KL, Gladden AD, Battis L, Kemper M, Zeng Q, Shea TP, Gujja S, Zedlack C, Gasser O. et al.Whole Genome Deep Sequencing of HIV-1 Reveals the Impact of Early Minor Variants Upon Immune Recognition During Acute Infection. PLoS Pathogens. 2012;8(3):e1002529. doi: 10.1371/journal.ppat.1002529. - DOI - PMC - PubMed
-
- Herbeck JT, Rolland M, Liu Y, McLaughlin S, McNevin J, Zhao H, Wong K, Stoddard JN, Raugi D, Sorensen S, Genowati I, Birditt B, McKay A, Diem K, Maust BS, Deng W, Collier AC, Stekler JD, McElrath MJ, Mullins JI. Demographic processes affect HIV-1 evolution in primary infection before the onset of selective processes. J Virol. 2011;85(15):7523–7534. doi: 10.1128/JVI.02697-10. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
