

An in vivo C. elegans model system for screening EGFR-inhibiting anti-cancer drugs
- PMID: 22957020
- PMCID: PMC3434183
- DOI: 10.1371/journal.pone.0042441
An in vivo C. elegans model system for screening EGFR-inhibiting anti-cancer drugs
Abstract
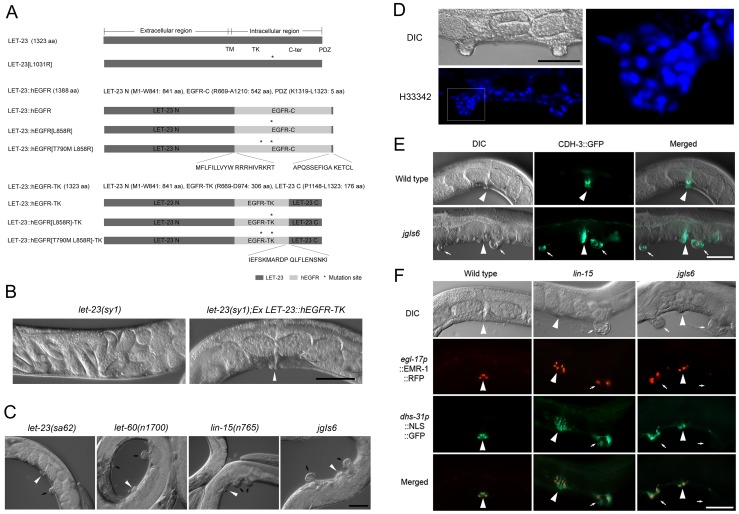
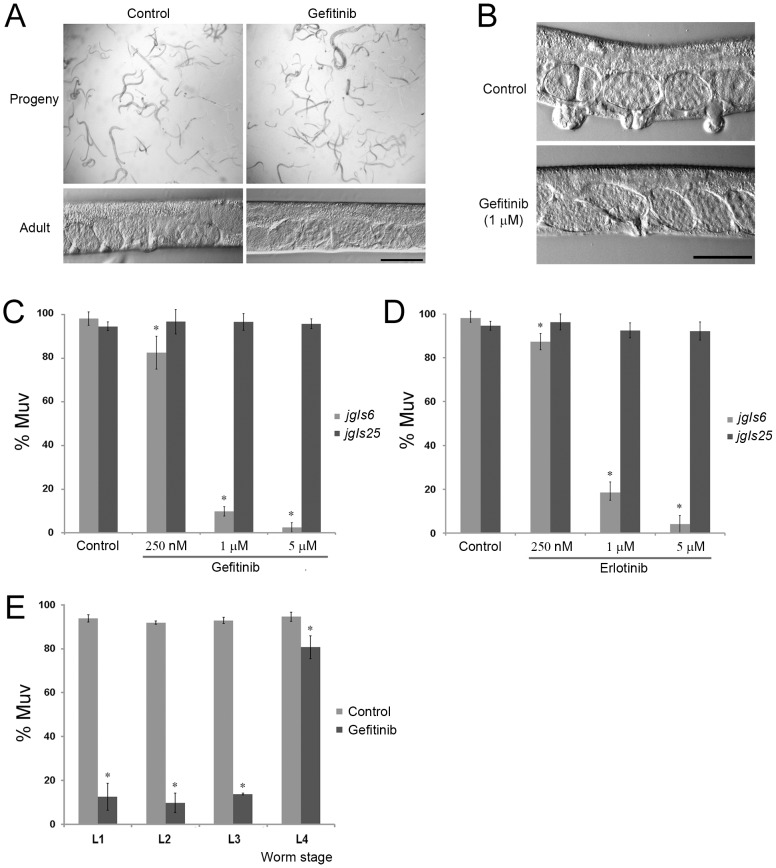
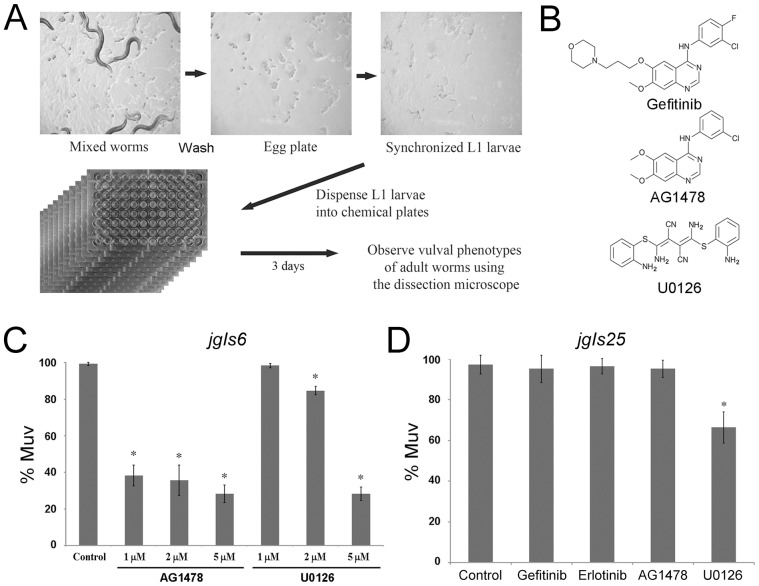
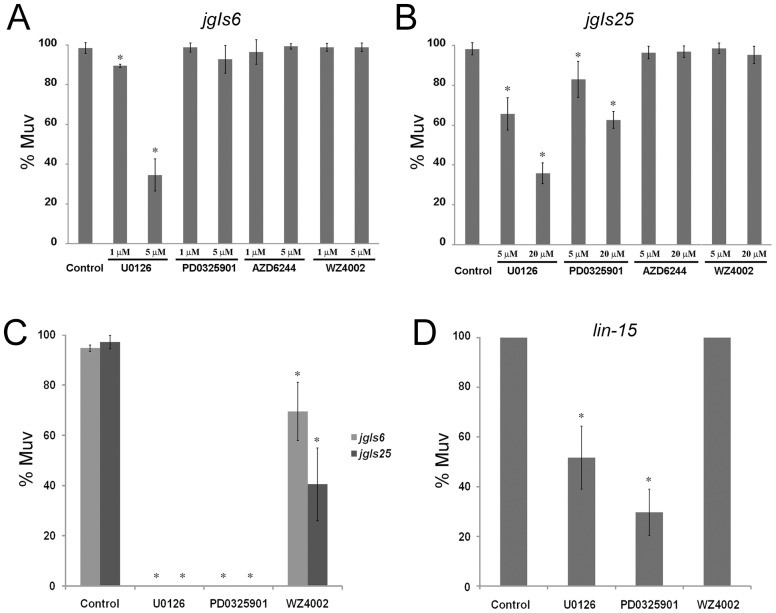
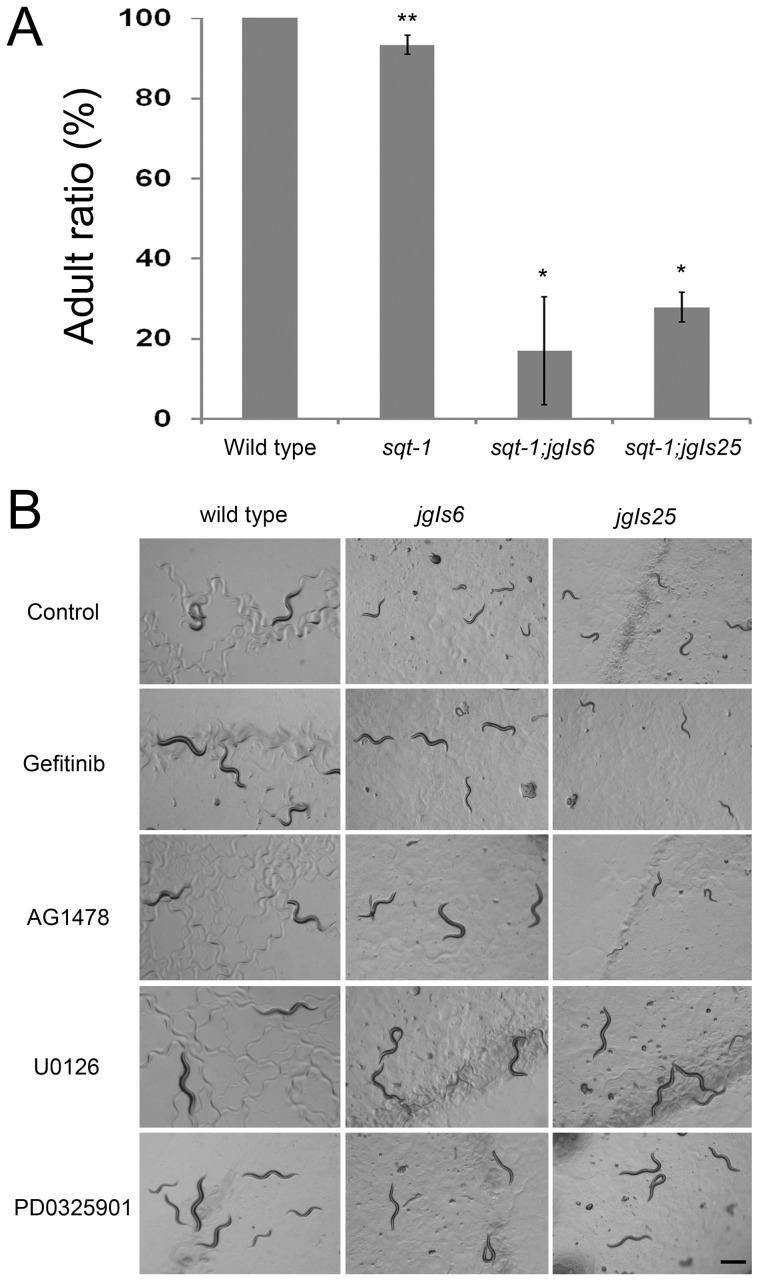
The epidermal growth factor receptor (EGFR) is a well-established target for cancer treatment. EGFR tyrosine kinase (TK) inhibitors, such as gefinitib and erlotinib, have been developed as anti-cancer drugs. Although non-small cell lung carcinoma with an activating EGFR mutation, L858R, responds well to gefinitib and erlotinib, tumors with a doubly mutated EGFR, T790M-L858R, acquire resistance to these drugs. The C. elegans EGFR homolog LET-23 and its downstream signaling pathway have been studied extensively to provide insight into regulatory mechanisms conserved from C. elegans to humans. To develop an in vivo screening system for potential cancer drugs targeting specific EGFR mutants, we expressed three LET-23 chimeras in which the TK domain was replaced with either the human wild-type TK domain (LET-23::hEGFR-TK), a TK domain with the L858R mutation (LET-23::hEGFR-TK[L858R]), or a TK domain with the T790M-L858R mutations (LET-23::hEGFR-TK[T790M-L858R]) in C. elegans vulval cells using the let-23 promoter. The wild-type hEGFR-TK chimeric protein rescued the let-23 mutant phenotype, and the activating mutant hEGFR-TK chimeras induced a multivulva (Muv) phenotype in a wild-type C. elegans background. The anti-cancer drugs gefitinib and erlotinib suppressed the Muv phenotype in LET-23::hEGFR-TK[L858R]-expressing transgenic animals, but not in LET-23::hEGFR-TK[T790M-L858R] transgenic animals. As a pilot screen, 8,960 small chemicals were tested for Muv suppression, and AG1478 (an EGFR-TK inhibitor) and U0126 (a MEK inhibitor) were identified as potential inhibitors of EGFR-mediated biological function. In conclusion, transgenic C. elegans expressing chimeric LET-23::hEGFR-TK proteins are a model system that can be used in mutation-specific screens for new anti-cancer drugs.
Conflict of interest statement
Figures
Similar articles
-
Structural basis for the altered drug sensitivities of non-small cell lung cancer-associated mutants of human epidermal growth factor receptor.Oncogene. 2013 Jan 3;32(1):27-38. doi: 10.1038/onc.2012.21. Epub 2012 Feb 20. Oncogene. 2013. PMID: 22349823
-
Rab8 and Rabin8-Mediated Tumor Formation by Hyperactivated EGFR Signaling via FGFR Signaling.Int J Mol Sci. 2020 Oct 20;21(20):7770. doi: 10.3390/ijms21207770. Int J Mol Sci. 2020. PMID: 33092268 Free PMC article.
-
Double EGFR mutants containing rare EGFR mutant types show reduced in vitro response to gefitinib compared with common activating missense mutations.Mol Cancer Ther. 2009 Aug;8(8):2142-51. doi: 10.1158/1535-7163.MCT-08-1219. Epub 2009 Aug 11. Mol Cancer Ther. 2009. PMID: 19671738
-
Activating and resistance mutations of EGFR in non-small-cell lung cancer: role in clinical response to EGFR tyrosine kinase inhibitors.Oncogene. 2009 Aug;28 Suppl 1(Suppl 1):S24-31. doi: 10.1038/onc.2009.198. Oncogene. 2009. PMID: 19680293 Free PMC article. Review.
-
Acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancers dependent on the epidermal growth factor receptor pathway.Clin Lung Cancer. 2009 Jul;10(4):281-9. doi: 10.3816/CLC.2009.n.039. Clin Lung Cancer. 2009. PMID: 19632948 Free PMC article. Review.
Cited by
-
Harmine suppresses hyper-activated Ras-MAPK pathway by selectively targeting oncogenic mutated Ras/Raf in Caenorhabditis elegans.Cancer Cell Int. 2019 Jun 11;19:159. doi: 10.1186/s12935-019-0880-4. eCollection 2019. Cancer Cell Int. 2019. PMID: 31198408 Free PMC article.
-
A survey of the kinome pharmacopeia reveals multiple scaffolds and targets for the development of novel anthelmintics.Sci Rep. 2021 Apr 28;11(1):9161. doi: 10.1038/s41598-021-88150-6. Sci Rep. 2021. PMID: 33911106 Free PMC article.
-
An approach using Caenorhabditis elegans screening novel targets to suppress tumour cell proliferation.Cell Prolif. 2020 Jun;53(6):e12832. doi: 10.1111/cpr.12832. Epub 2020 May 25. Cell Prolif. 2020. PMID: 32452127 Free PMC article.
-
Isoharringtonine Induces Apoptosis of Non-Small Cell Lung Cancer Cells in Tumorspheroids via the Intrinsic Pathway.Biomolecules. 2020 Nov 6;10(11):1521. doi: 10.3390/biom10111521. Biomolecules. 2020. PMID: 33172112 Free PMC article.
-
A Drug Discovery Pipeline for MAPK/ERK Pathway Inhibitors in Caenorhabditis elegans.Cancer Res Commun. 2024 Sep 1;4(9):2454-2462. doi: 10.1158/2767-9764.CRC-24-0221. Cancer Res Commun. 2024. PMID: 39212544 Free PMC article.
References
-
- Artal-Sanz M, de Jong L, Tavernarakis N (2006) Caenorhabditis elegans: a versatile platform for drug discovery. Biotechnol J 1: 1405–1418. - PubMed
-
- Hynes NE, Lane HA (2005) ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer 5: 341–354. - PubMed
-
- Gschwind A, Fischer OM, Ullrich A (2004) The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat Rev Cancer 4: 361–370. - PubMed
-
- Muhsin M, Graham J, Kirkpatrick P (2003) Gefitinib. Nat Rev Drug Discov 2: 515–516. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
