

Transcriptional regulation of the operon encoding stress-responsive ECF sigma factor SigH and its anti-sigma factor RshA, and control of its regulatory network in Corynebacterium glutamicum
- PMID: 22943411
- PMCID: PMC3489674
- DOI: 10.1186/1471-2164-13-445
Transcriptional regulation of the operon encoding stress-responsive ECF sigma factor SigH and its anti-sigma factor RshA, and control of its regulatory network in Corynebacterium glutamicum
Abstract
Background: The expression of genes in Corynebacterium glutamicum, a Gram-positive non-pathogenic bacterium used mainly for the industrial production of amino acids, is regulated by seven different sigma factors of RNA polymerase, including the stress-responsive ECF-sigma factor SigH. The sigH gene is located in a gene cluster together with the rshA gene, putatively encoding an anti-sigma factor. The aim of this study was to analyze the transcriptional regulation of the sigH and rshA gene cluster and the effects of RshA on the SigH regulon, in order to refine the model describing the role of SigH and RshA during stress response.
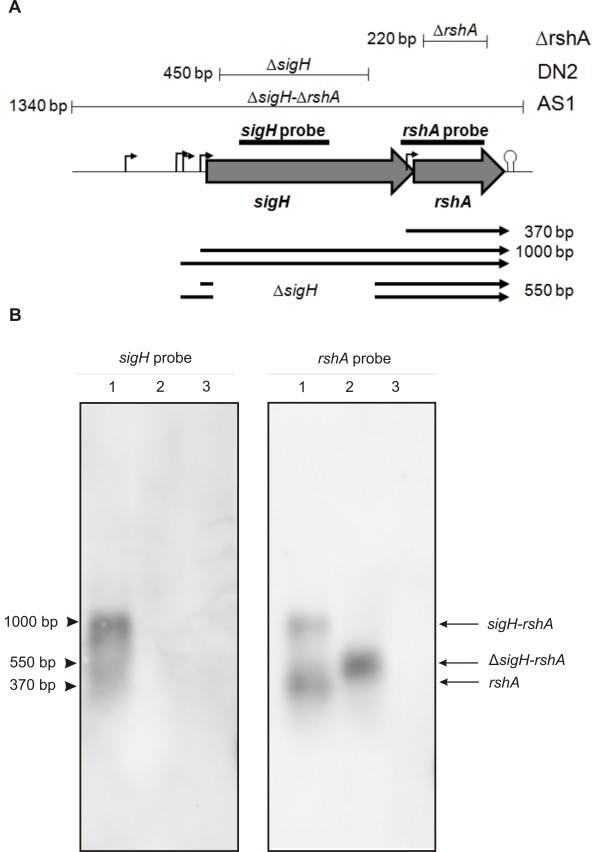
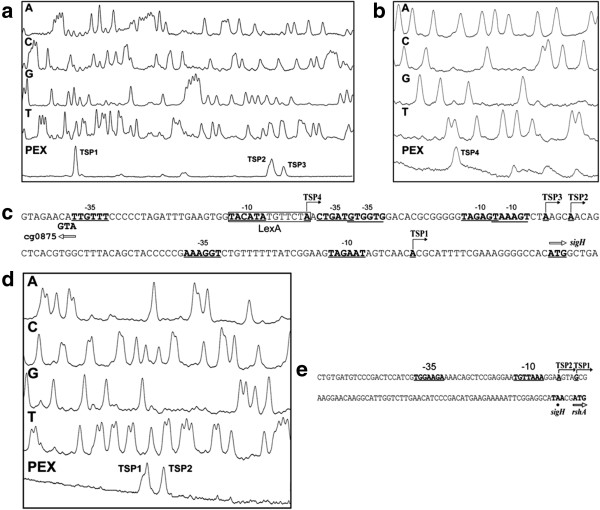
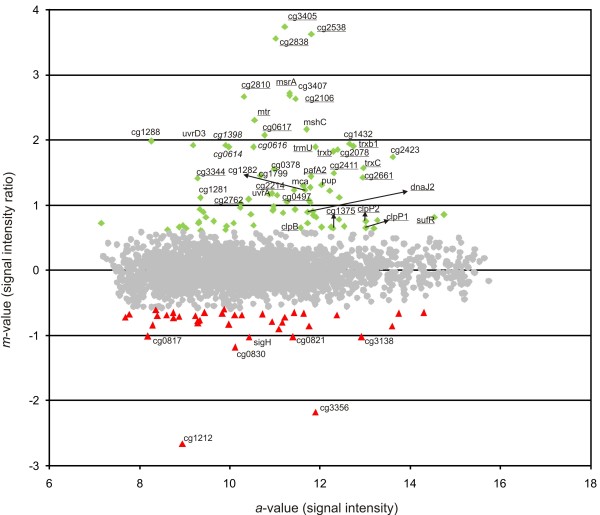
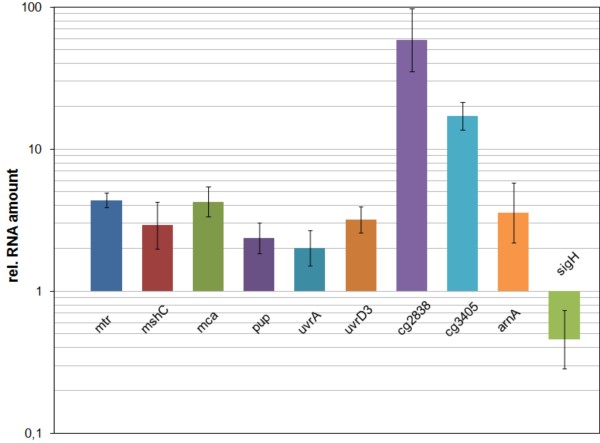
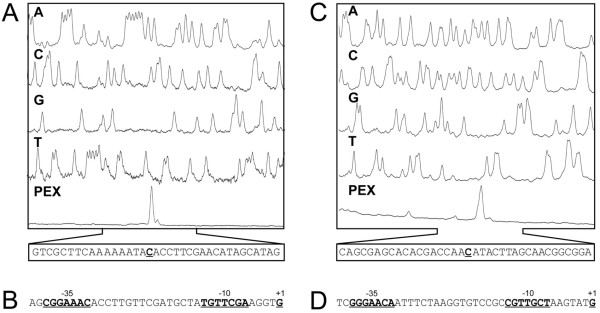
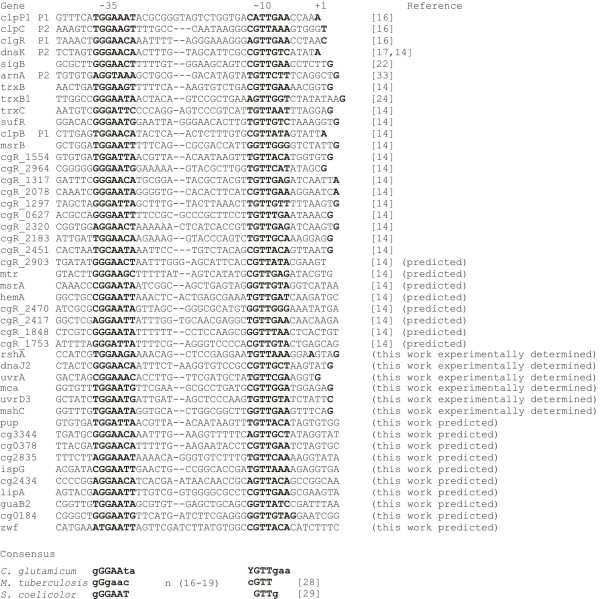
Results: Transcription analyses revealed that the sigH gene and rshA gene are cotranscribed from four sigH housekeeping promoters in C. glutamicum. In addition, a SigH-controlled rshA promoter was found to only drive the transcription of the rshA gene. To test the role of the putative anti-sigma factor gene rshA under normal growth conditions, a C. glutamicum rshA deletion strain was constructed and used for genome-wide transcription profiling with DNA microarrays. In total, 83 genes organized in 61 putative transcriptional units, including those previously detected using sigH mutant strains, exhibited increased transcript levels in the rshA deletion mutant compared to its parental strain. The genes encoding proteins related to disulphide stress response, heat stress proteins, components of the SOS-response to DNA damage and proteasome components were the most markedly upregulated gene groups. Altogether six SigH-dependent promoters upstream of the identified genes were determined by primer extension and a refined consensus promoter consisting of 45 original promoter sequences was constructed.
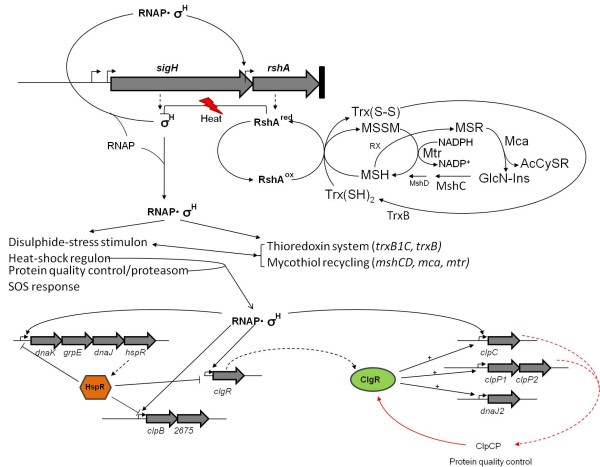
Conclusions: The rshA gene codes for an anti-sigma factor controlling the function of the stress-responsive sigma factor SigH in C. glutamicum. Transcription of rshA from a SigH-dependent promoter may serve to quickly shutdown the SigH-dependent stress response after the cells have overcome the stress condition. Here we propose a model of the regulation of oxidative and heat stress response including redox homeostasis by SigH, RshA and the thioredoxin system.
Figures

Similar articles
-
The extracytoplasmic function-type sigma factor SigM of Corynebacterium glutamicum ATCC 13032 is involved in transcription of disulfide stress-related genes.J Bacteriol. 2007 Jul;189(13):4696-707. doi: 10.1128/JB.00382-07. Epub 2007 May 4. J Bacteriol. 2007. PMID: 17483229 Free PMC article.
-
Expanding the regulatory network governed by the extracytoplasmic function sigma factor σH in Corynebacterium glutamicum.J Bacteriol. 2015 Feb;197(3):483-96. doi: 10.1128/JB.02248-14. Epub 2014 Nov 17. J Bacteriol. 2015. PMID: 25404703 Free PMC article.
-
Regulation of Corynebacterium glutamicum heat shock response by the extracytoplasmic-function sigma factor SigH and transcriptional regulators HspR and HrcA.J Bacteriol. 2009 May;191(9):2964-72. doi: 10.1128/JB.00112-09. Epub 2009 Mar 6. J Bacteriol. 2009. PMID: 19270092 Free PMC article.
-
Sigma factors and promoters in Corynebacterium glutamicum.J Biotechnol. 2011 Jul 10;154(2-3):101-13. doi: 10.1016/j.jbiotec.2011.01.017. Epub 2011 Jan 26. J Biotechnol. 2011. PMID: 21277915 Review.
-
Analysis of Corynebacterium glutamicum promoters and their applications.Subcell Biochem. 2012;64:203-21. doi: 10.1007/978-94-007-5055-5_10. Subcell Biochem. 2012. PMID: 23080252 Review.
Cited by
-
Physiological roles of sigma factor SigD in Corynebacterium glutamicum.BMC Microbiol. 2017 Jul 12;17(1):158. doi: 10.1186/s12866-017-1067-6. BMC Microbiol. 2017. PMID: 28701150 Free PMC article.
-
A novel mycothiol-dependent thiol-disulfide reductase in Corynebacterium glutamicum involving oxidative stress resistance.3 Biotech. 2021 Aug;11(8):372. doi: 10.1007/s13205-021-02896-4. Epub 2021 Jul 14. 3 Biotech. 2021. PMID: 34290951 Free PMC article.
-
Characterization of Sigma Factor Genes in Streptomyces lividans TK24 Using a Genomic Library-Based Approach for Multiple Gene Deletions.Front Microbiol. 2018 Dec 10;9:3033. doi: 10.3389/fmicb.2018.03033. eCollection 2018. Front Microbiol. 2018. PMID: 30619125 Free PMC article.
-
Secretome Dynamics in a Gram-Positive Bacterial Model.Mol Cell Proteomics. 2019 Mar;18(3):423-436. doi: 10.1074/mcp.RA118.000899. Epub 2018 Nov 29. Mol Cell Proteomics. 2019. PMID: 30498012 Free PMC article.
-
Identification of Rhodococcus erythropolis Promoters Controlled by Alternative Sigma Factors Using In Vivo and In Vitro Systems and Heterologous RNA Polymerase.Curr Microbiol. 2022 Jan 4;79(2):55. doi: 10.1007/s00284-021-02747-8. Curr Microbiol. 2022. PMID: 34982253
References
-
- Brune I, Brinkrolf K, Kalinowski J, Pühler A, Tauch A. The individual and common repertoire of DNA-binding transcriptional regulators of Corynebacterium glutamicum, Corynebacterium efficiens, Corynebacterium diphtheriae and Corynebacterium jeikeium deduced from the complete genome sequences. BMC Genom. 2005;6:86. doi: 10.1186/1471-2164-6-86. - DOI - PMC - PubMed
-
- Mishra AK, Alderwick LJ, Rittmann D, Tatituri RV, Nigou J, Gilleron M, Eggeling L, Besra GS. Identification of an alpha(1→6) mannopyranosyltransferase (MptA), involved in Corynebacterium glutamicum lipomanann biosynthesis, and identification of its orthologue in Mycobacterium tuberculosis. Mol Microbiol. 2007;65:1503–1517. doi: 10.1111/j.1365-2958.2007.05884.x. - DOI - PMC - PubMed
-
- Möker N, Brocker M, Schaffer S, Krämer R, Morbach S, Bott M. Deletion of the genes encoding the MtrA-MtrB two-component system of Corynebacterium glutamicum has a strong influence on cell morphology, antibiotics susceptibility and expression of genes involved in osmoprotection. Mol Microbiol. 2004;54:420–438. doi: 10.1111/j.1365-2958.2004.04249.x. - DOI - PubMed
-
- Kalinowski J, Bathe B, Bartels D, Bischoff N, Bott M, Burkovski A, Dusch N, Eggeling L, Eikmanns BJ, Gaigalat L, Goesmann A, Hartmann M, Huthmacher K, Krämer R, Linke B, McHardy AC, Meyer F, Möckel B, Pfefferle W, Pühler A, Rey DA, Ruckert C, Rupp O, Sahm H, Wendisch VF, Wiegräbe I, Tauch A. The complete Corynebacterium glutamicum ATCC 13032 genome sequence and its impact on the production of L-aspartate-derived amino acids and vitamins. J Biotechnol. 2003;104:5–25. doi: 10.1016/S0168-1656(03)00154-8. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
