

Myo1c binding to submembrane actin mediates insulin-induced tethering of GLUT4 vesicles
- PMID: 22918957
- PMCID: PMC3469521
- DOI: 10.1091/mbc.E12-04-0263
Myo1c binding to submembrane actin mediates insulin-induced tethering of GLUT4 vesicles
Abstract
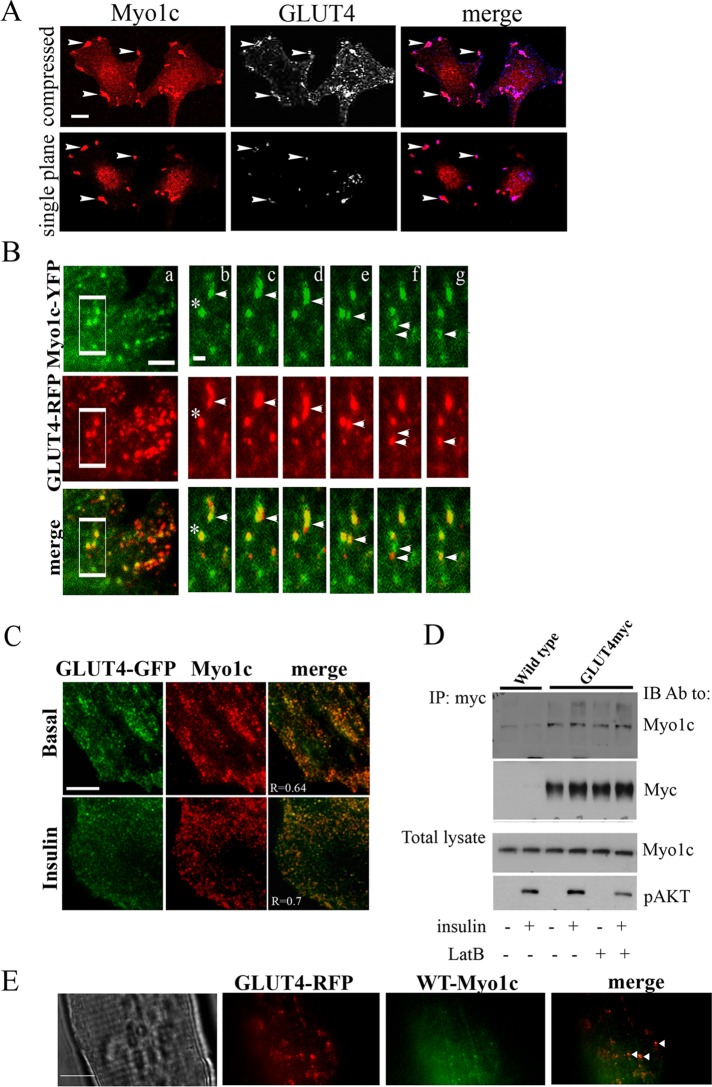
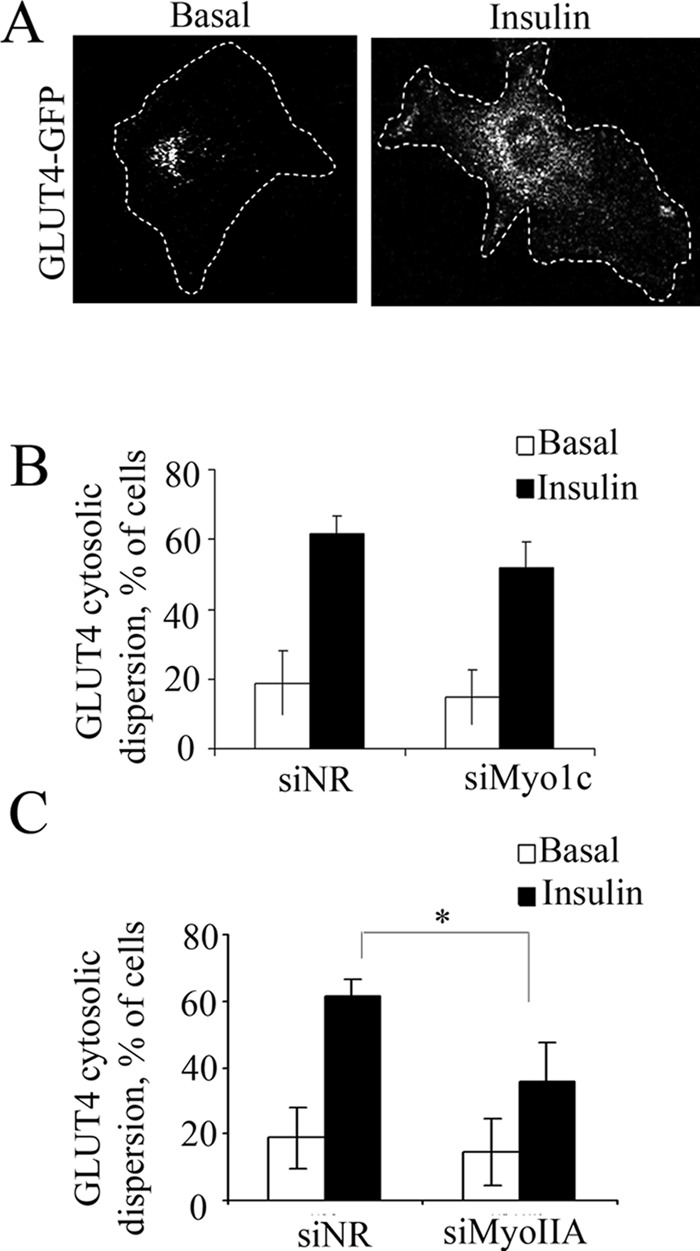
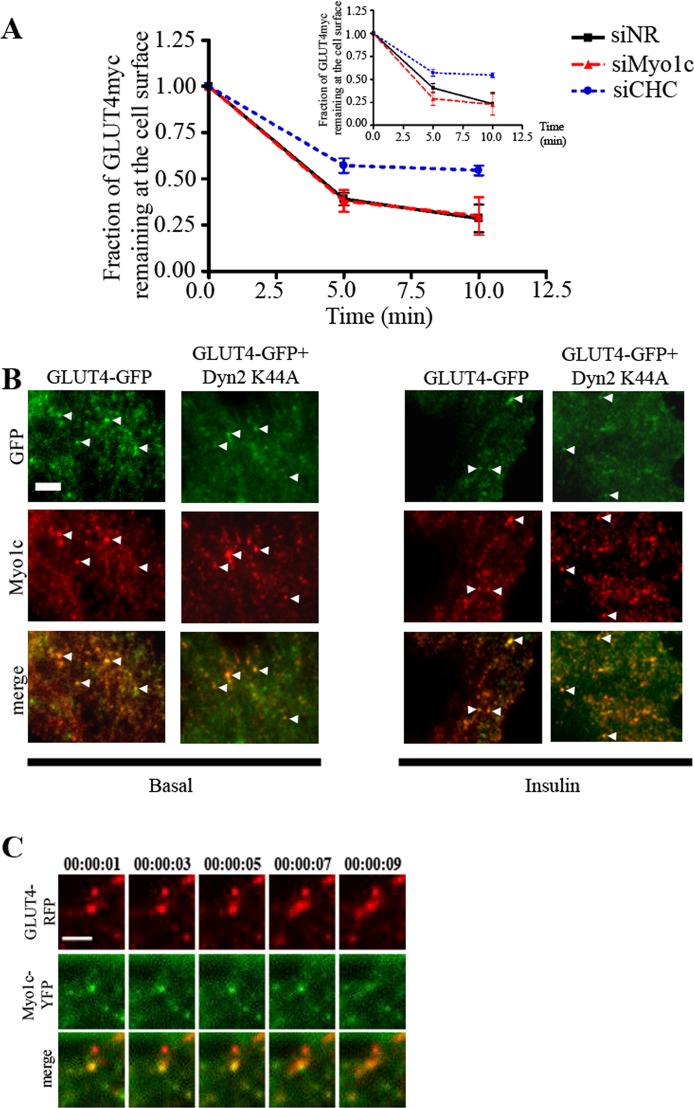
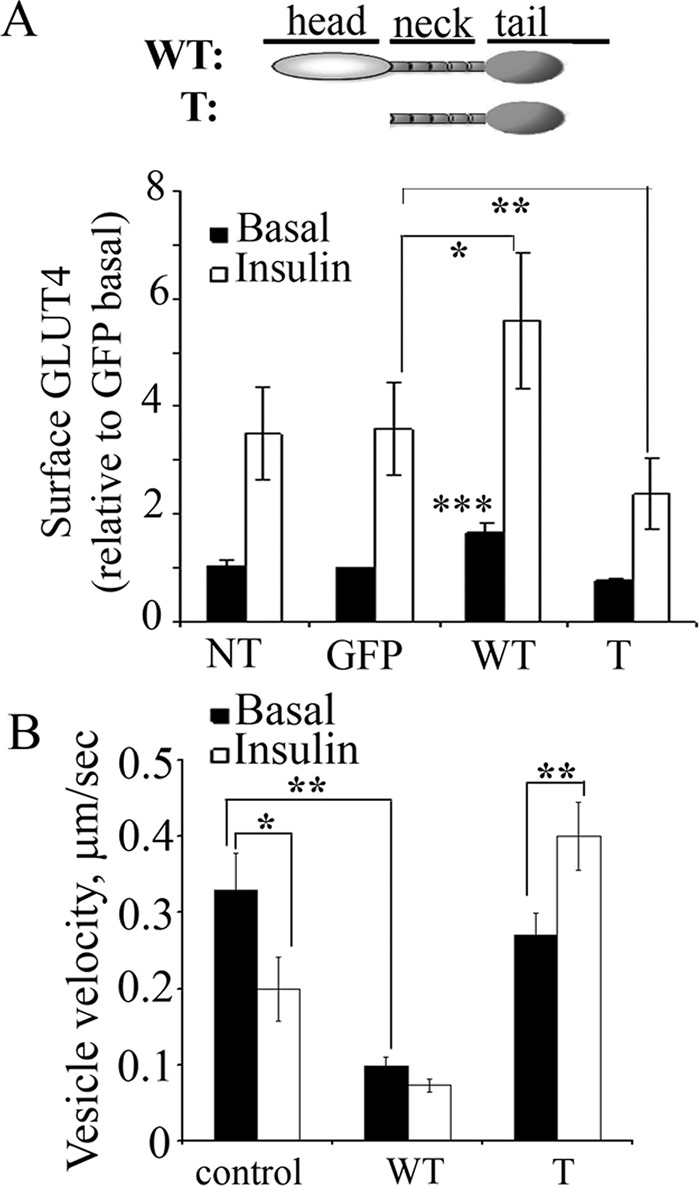
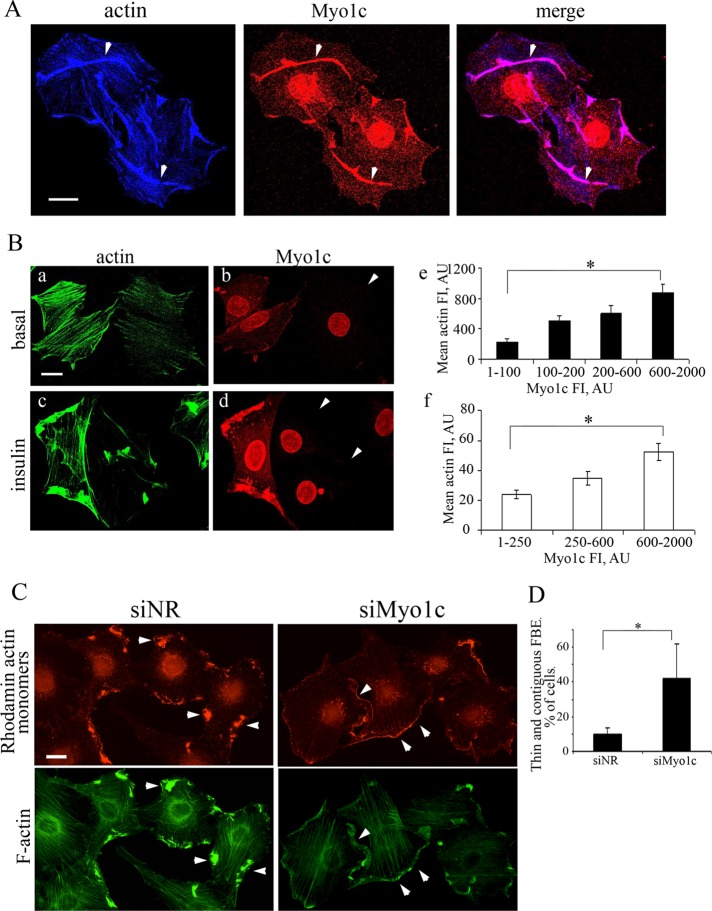
GLUT4-containing vesicles cycle between the plasma membrane and intracellular compartments. Insulin promotes GLUT4 exocytosis by regulating GLUT4 vesicle arrival at the cell periphery and its subsequent tethering, docking, and fusion with the plasma membrane. The molecular machinery involved in GLUT4 vesicle tethering is unknown. We show here that Myo1c, an actin-based motor protein that associates with membranes and actin filaments, is required for insulin-induced vesicle tethering in muscle cells. Myo1c was found to associate with both mobile and tethered GLUT4 vesicles and to be required for vesicle capture in the total internal reflection fluorescence (TIRF) zone beneath the plasma membrane. Myo1c knockdown or overexpression of an actin binding-deficient Myo1c mutant abolished insulin-induced vesicle immobilization, increased GLUT4 vesicle velocity in the TIRF zone, and prevented their externalization. Conversely, Myo1c overexpression immobilized GLUT4 vesicles in the TIRF zone and promoted insulin-induced GLUT4 exposure to the extracellular milieu. Myo1c also contributed to insulin-dependent actin filament remodeling. Thus we propose that interaction of vesicular Myo1c with cortical actin filaments is required for insulin-mediated tethering of GLUT4 vesicles and for efficient GLUT4 surface delivery in muscle cells.
Figures

Similar articles
-
Update on GLUT4 Vesicle Traffic: A Cornerstone of Insulin Action.Trends Endocrinol Metab. 2017 Aug;28(8):597-611. doi: 10.1016/j.tem.2017.05.002. Epub 2017 Jun 8. Trends Endocrinol Metab. 2017. PMID: 28602209 Review.
-
Activation of RalA is required for insulin-stimulated Glut4 trafficking to the plasma membrane via the exocyst and the motor protein Myo1c.Dev Cell. 2007 Sep;13(3):391-404. doi: 10.1016/j.devcel.2007.07.007. Dev Cell. 2007. PMID: 17765682
-
Myosin Va mediates Rab8A-regulated GLUT4 vesicle exocytosis in insulin-stimulated muscle cells.Mol Biol Cell. 2014 Apr;25(7):1159-70. doi: 10.1091/mbc.E13-08-0493. Epub 2014 Jan 29. Mol Biol Cell. 2014. PMID: 24478457 Free PMC article.
-
Insulin stimulates membrane fusion and GLUT4 accumulation in clathrin coats on adipocyte plasma membranes.Mol Cell Biol. 2007 May;27(9):3456-69. doi: 10.1128/MCB.01719-06. Epub 2007 Mar 5. Mol Cell Biol. 2007. PMID: 17339344 Free PMC article.
-
GLUT4 translocation: the last 200 nanometers.Cell Signal. 2007 Nov;19(11):2209-17. doi: 10.1016/j.cellsig.2007.06.003. Epub 2007 Jun 21. Cell Signal. 2007. PMID: 17629673 Review.
Cited by
-
Myosin-I molecular motors at a glance.J Cell Sci. 2016 Jul 15;129(14):2689-95. doi: 10.1242/jcs.186403. Epub 2016 Jul 11. J Cell Sci. 2016. PMID: 27401928 Free PMC article. Review.
-
Structure of myosin-1c tail bound to calmodulin provides insights into calcium-mediated conformational coupling.Nat Struct Mol Biol. 2015 Jan;22(1):81-8. doi: 10.1038/nsmb.2923. Epub 2014 Dec 1. Nat Struct Mol Biol. 2015. PMID: 25437912
-
Promoting Glucose Transporter-4 Vesicle Trafficking along Cytoskeletal Tracks: PAK-Ing Them Out.Front Endocrinol (Lausanne). 2017 Nov 20;8:329. doi: 10.3389/fendo.2017.00329. eCollection 2017. Front Endocrinol (Lausanne). 2017. PMID: 29209279 Free PMC article. Review.
-
An analysis of DNA methylation in human adipose tissue reveals differential modification of obesity genes before and after gastric bypass and weight loss.Genome Biol. 2015 Jan 22;16(1):8. doi: 10.1186/s13059-014-0569-x. Genome Biol. 2015. PMID: 25651499 Free PMC article.
-
The motor protein Myo1c regulates transforming growth factor-β-signaling and fibrosis in podocytes.Kidney Int. 2019 Jul;96(1):139-158. doi: 10.1016/j.kint.2019.02.014. Epub 2019 Mar 4. Kidney Int. 2019. PMID: 31097328 Free PMC article.
References
-
- Adams RJ, Pollard TD. Binding of myosin I to membrane lipids. Nature. 1989;340:565–568. - PubMed
-
- Antonescu CN, Diaz M, Femia G, Planas JV, Klip A. Clathrin-dependent and independent endocytosis of glucose transporter 4 (GLUT4) in myoblasts: regulation by mitochondrial uncoupling. Traffic. 2008;9:1173–1190. - PubMed
-
- Barylko B, Jung G, Albanesi JP. Structure, function, and regulation of myosin 1C. Acta Biochim Pol. 2005;52:373–380. - PubMed
-
- Bayer KU, De Koninck P, Leonard AS, Hell JW, Schulman H. Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature. 2001;411:801–805. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
