

Protein kinases display minimal interpositional dependence on substrate sequence: potential implications for the evolution of signalling networks
- PMID: 22889908
- PMCID: PMC3415838
- DOI: 10.1098/rstb.2012.0010
Protein kinases display minimal interpositional dependence on substrate sequence: potential implications for the evolution of signalling networks
Abstract
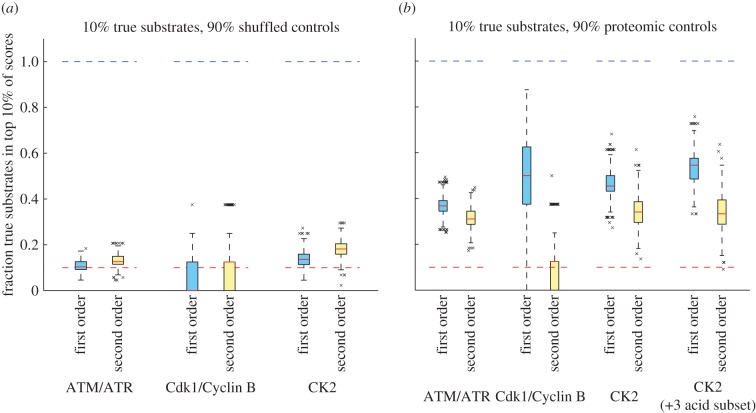
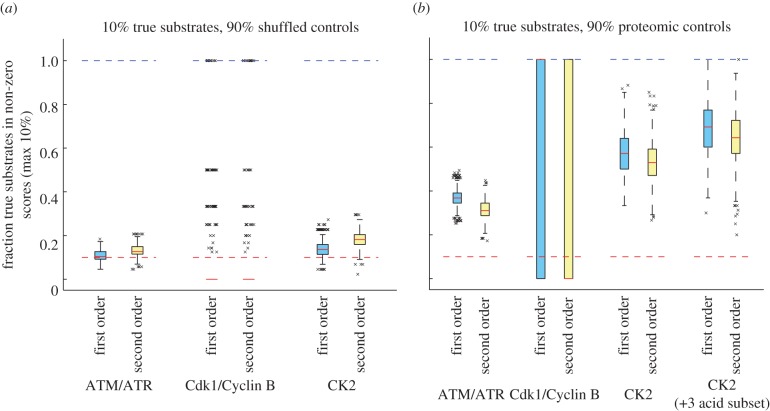
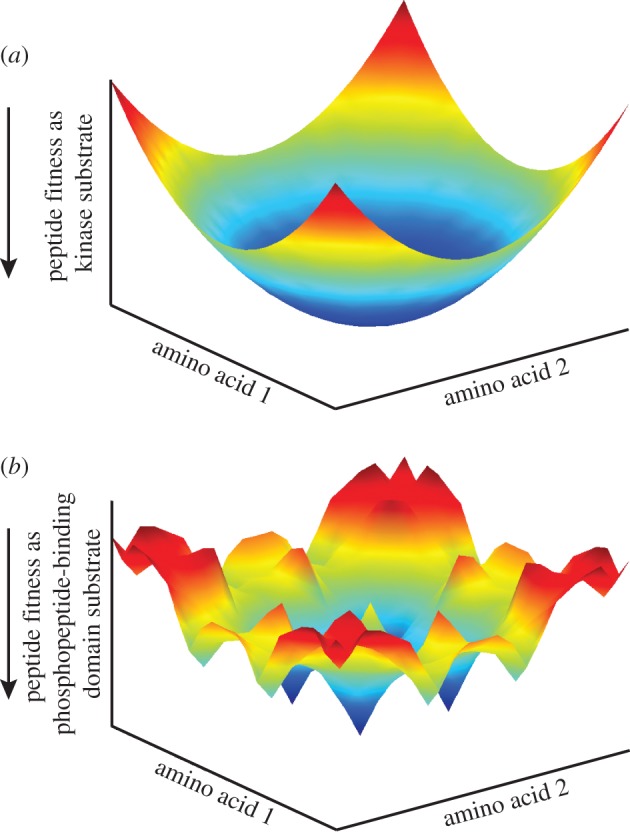
Characterization of in vitro substrates of protein kinases by peptide library screening provides a wealth of information on the substrate specificity of kinases for amino acids at particular positions relative to the site of phosphorylation, but provides no information concerning interdependence among positions. High-throughput techniques have recently made it feasible to identify large numbers of in vivo kinase substrates. We used data from experiments on the kinases ATM/ATR and CDK1, and curated CK2 substrates to evaluate the prevalence of interactions between substrate positions within a motif and the utility of these interactions in predicting kinase substrates. Among these data, evidence of interpositional sequence dependencies is strikingly rare, and what dependency exists does little to aid in the prediction of novel kinase substrates. Significant increases in the ability of models to predict kinase-substrate specificity beyond position-independent models must come largely from inclusion of elements of biological and cellular context, rather than further analysis of substrate sequences alone. Our results suggest that, evolutionarily, kinase substrate fitness exists in a smooth energetic landscape. Taken with results from others indicating that phosphopeptide-binding domains do exhibit interpositional dependence, our data suggest that incorporation of new substrate molecules into phospho-signalling networks may be rate-limited by the evolution of suitability for binding by phosphopeptide-binding domains.
Figures
Similar articles
-
Utilization of oriented peptide libraries to identify substrate motifs selected by ATM.J Biol Chem. 2000 Jul 28;275(30):22719-27. doi: 10.1074/jbc.M001002200. J Biol Chem. 2000. PMID: 10801797
-
The Wip1 phosphatase PPM1D dephosphorylates SQ/TQ motifs in checkpoint substrates phosphorylated by PI3K-like kinases.Biochemistry. 2007 Nov 6;46(44):12594-603. doi: 10.1021/bi701096s. Epub 2007 Oct 16. Biochemistry. 2007. PMID: 17939684
-
The molecular basis of FHA domain:phosphopeptide binding specificity and implications for phospho-dependent signaling mechanisms.Mol Cell. 2000 Nov;6(5):1169-82. doi: 10.1016/s1097-2765(00)00114-3. Mol Cell. 2000. PMID: 11106755
-
Recognition and specificity in protein tyrosine kinase-mediated signalling.Trends Biochem Sci. 1995 Nov;20(11):470-5. doi: 10.1016/s0968-0004(00)89103-3. Trends Biochem Sci. 1995. PMID: 8578591 Review.
-
SQ/TQ cluster domains: concentrated ATM/ATR kinase phosphorylation site regions in DNA-damage-response proteins.Bioessays. 2005 Apr;27(4):397-407. doi: 10.1002/bies.20204. Bioessays. 2005. PMID: 15770685 Review.
Cited by
-
Ancestral resurrection reveals evolutionary mechanisms of kinase plasticity.Elife. 2014 Oct 13;3:e04126. doi: 10.7554/eLife.04126. Elife. 2014. PMID: 25310241 Free PMC article.
-
Duck plague virus US3 protein kinase phosphorylates UL47 and regulates the subcellular localization of UL47.Front Microbiol. 2022 Oct 25;13:876820. doi: 10.3389/fmicb.2022.876820. eCollection 2022. Front Microbiol. 2022. PMID: 36386680 Free PMC article.
-
Exploiting holistic approaches to model specificity in protein phosphorylation.Front Genet. 2014 Sep 30;5:315. doi: 10.3389/fgene.2014.00315. eCollection 2014. Front Genet. 2014. PMID: 25324856 Free PMC article. Review.
-
Comprehensive substrate specificity profiling of the human Nek kinome reveals unexpected signaling outputs.Elife. 2019 May 24;8:e44635. doi: 10.7554/eLife.44635. Elife. 2019. PMID: 31124786 Free PMC article.
-
Regulation and function of phosphorylation on VP8, the major tegument protein of bovine herpesvirus 1.J Virol. 2015 Apr;89(8):4598-611. doi: 10.1128/JVI.03180-14. Epub 2015 Feb 11. J Virol. 2015. PMID: 25673708 Free PMC article.
References
-
- Amanchy R., Periaswamy B., Mathivanan S., Reddy R., Tattikota S. G., Pandey A. 2007. A curated compendium of phosphorylation motifs. Nat. Biotechnol. 25, 285–286 10.1038/nbt0307-285 (doi:10.1038/nbt0307-285) - DOI - DOI - PubMed
-
- Alexander J., et al. 2011. Spatial exclusivity combined with positive and negative selection of phosphorylation motifs is the basis for context-dependent mitotic signaling. Sci. Signal. 4, ra42 10.1126/scisignal.2001796 (doi:10.1126/scisignal.2001796) - DOI - DOI - PMC - PubMed
-
- Obenauer J. C., Cantley L. C., Yaffe M. B. 2003. Scansite 2.0: proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 31, 3635–3741 10.1093/nar/gkg584 (doi:10.1093/nar/gkg584) - DOI - DOI - PMC - PubMed
-
- Blom N., Sicheritz-Ponten T., Gupta R., Gammeltoft S., Brunak S. 2004. Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 4, 1633–1649 10.1002/pmic.200300771 (doi:10.1002/pmic.200300771) - DOI - DOI - PubMed
-
- Miller M. L., et al. 2008. Linear motif atlas for phosphorylation-dependent signaling. Sci Signal. 1, ra2 10.1126/scisignal.1159433 (doi:10.1126/scisignal.1159433) - DOI - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
