

E2F1 confers anticancer drug resistance by targeting ABC transporter family members and Bcl-2 via the p73/DNp73-miR-205 circuitry
- PMID: 22871739
- PMCID: PMC3442917
- DOI: 10.4161/cc.21476
E2F1 confers anticancer drug resistance by targeting ABC transporter family members and Bcl-2 via the p73/DNp73-miR-205 circuitry
Abstract
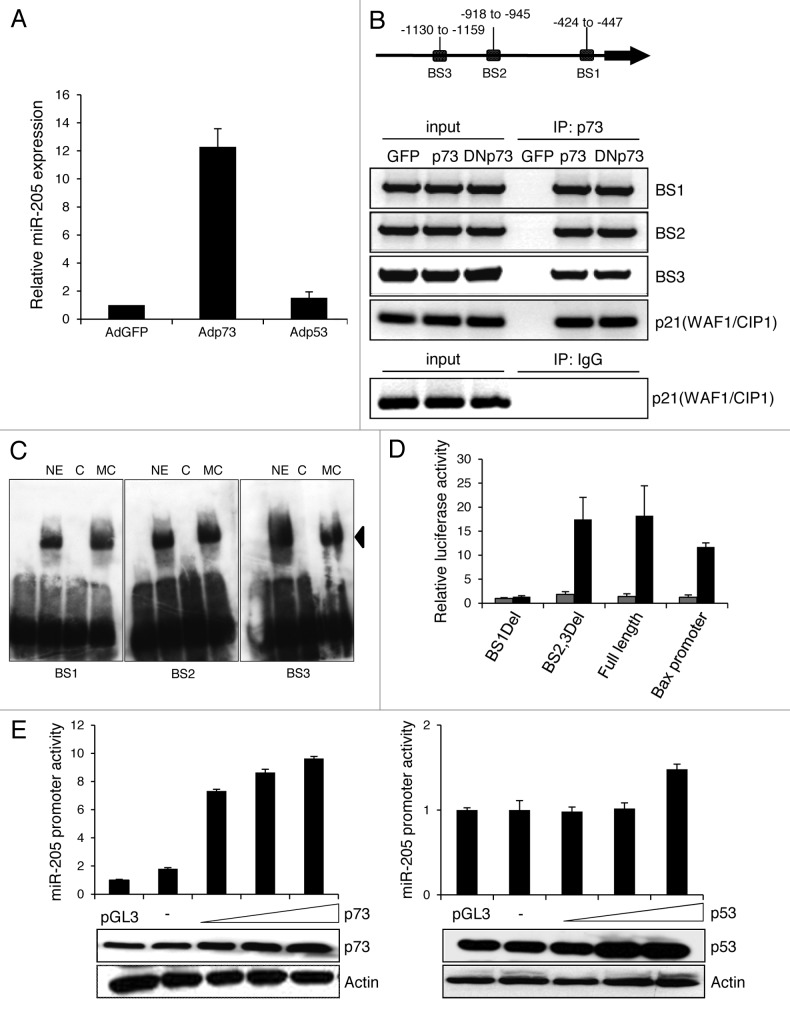
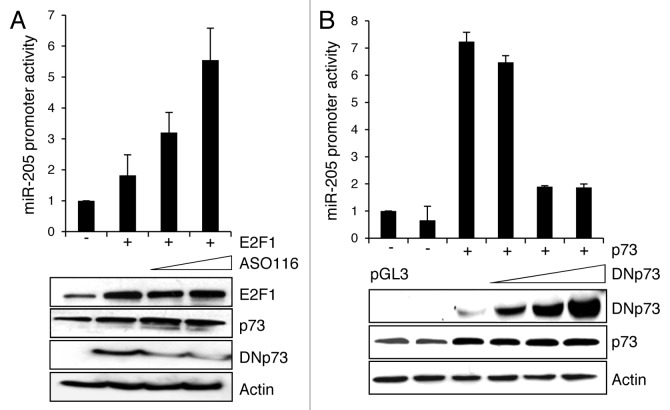
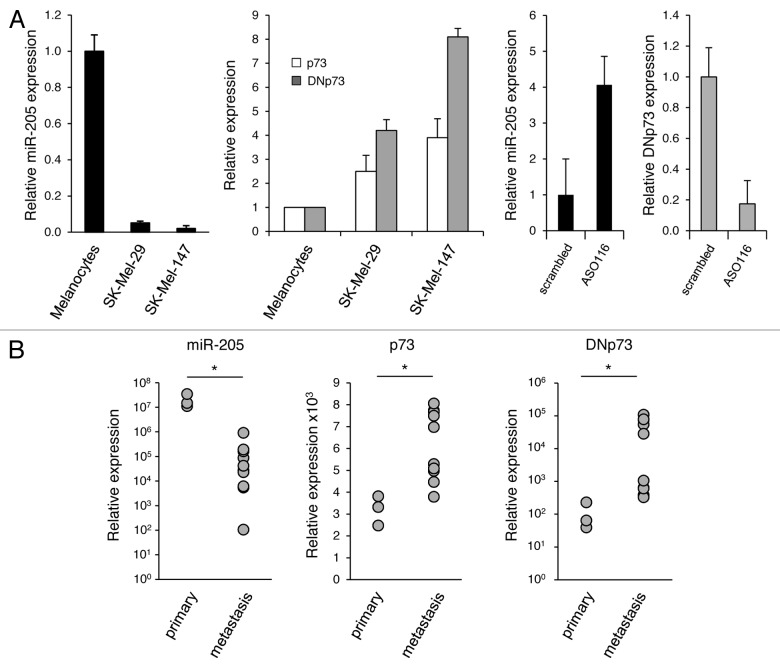
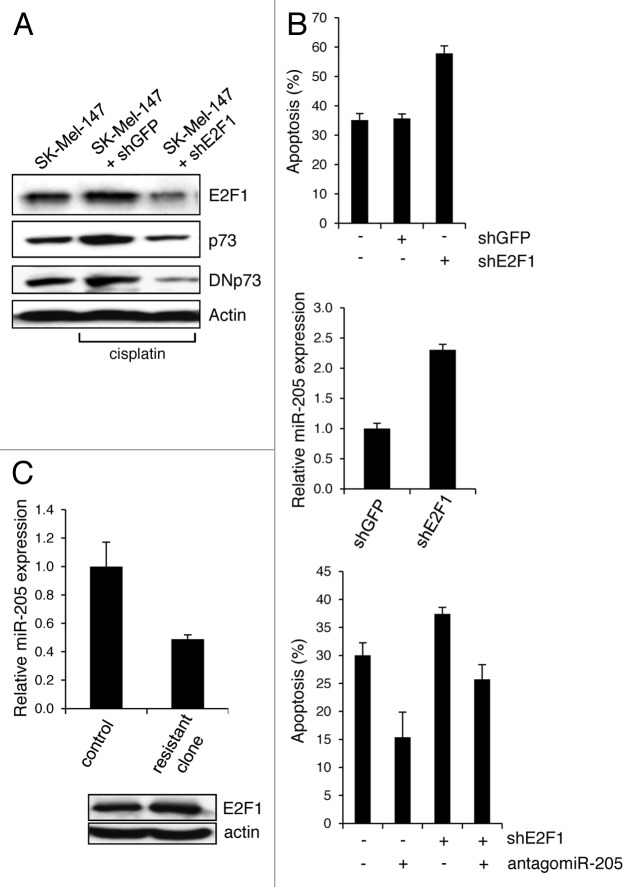
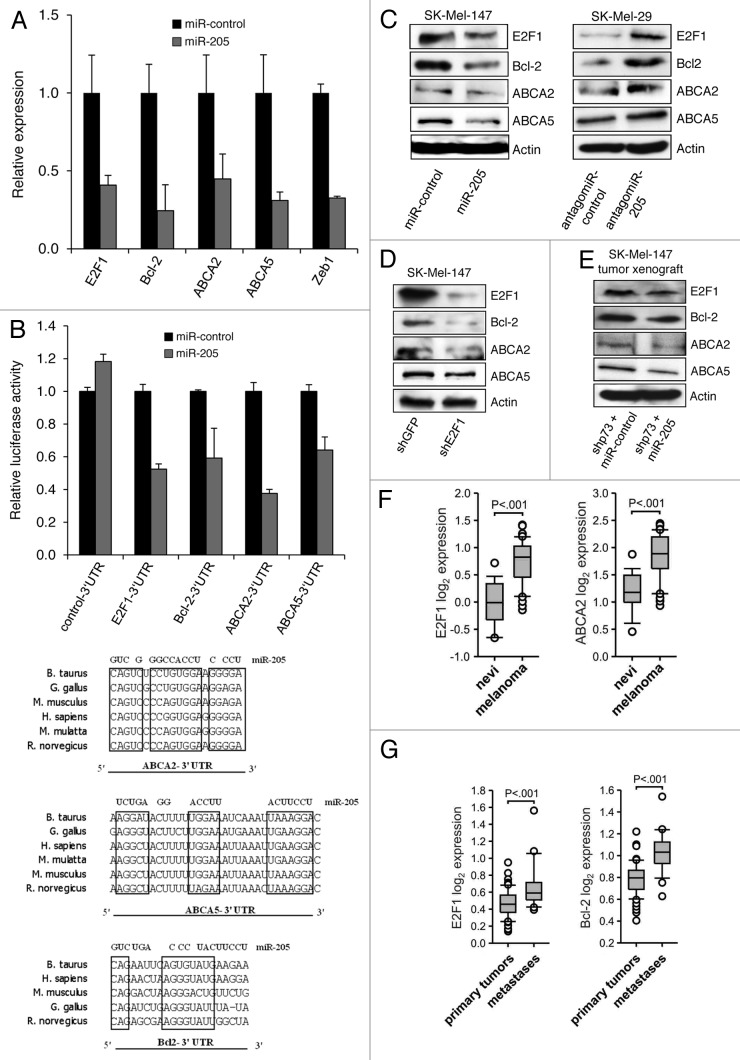
Resistance to anti-neoplastic agents is the major cause of therapy failure, leading to disease recurrence and metastasis. E2F1 is a strong inducer of apoptosis in response to DNA damage through its capacity to activate p53/p73 death pathways. Recent evidence, however, showed that E2F1, which is aberrantly expressed in advanced malignant melanomas together with antagonistic p73 family members, drives cancer progression. Investigating mechanisms responsible for dysregulated E2F1 losing its apoptotic function, we searched for genomic signatures in primary and late clinical tumor stages to allow the prediction of downstream effectors associated with apoptosis resistance and survival of aggressive melanoma cells. We identified miR-205 as specific target of p73 and found that upon genotoxic stress, its expression is sufficiently abrogated by endogenous DNp73. Significantly, metastatic cells can be rescued from drug resistance by selective knockdown of DNp73 or overexpression of miR-205 in p73-depleted cells, leading to increased apoptosis and the reduction of tumor growth in vivo. Our data delineate an autoregulatory circuit, involving high levels of E2F1 and DNp73 to downregulate miR-205, which, in turn, controls E2F1 accumulation. Finally, drug resistance associated to this genetic signature is mediated by removing the inhibitory effect of miR-205 on the expression of Bcl-2 and the ATP-binding cassette transporters A2 (ABCA2) and A5 (ABCA5) related to multi-drug resistance and malignant progression. These results define the E2F1-p73/DNp73-miR-205 axis as a crucial mechanism for chemoresistance and, thus, as a target for metastasis prevention.
Figures

Similar articles
-
Kinetic modeling-based detection of genetic signatures that provide chemoresistance via the E2F1-p73/DNp73-miR-205 network.Cancer Res. 2013 Jun 15;73(12):3511-24. doi: 10.1158/0008-5472.CAN-12-4095. Epub 2013 Feb 27. Cancer Res. 2013. PMID: 23447575
-
Mdm2 inhibition induces apoptosis in p53 deficient human colon cancer cells by activating p73- and E2F1-mediated expression of PUMA and Siva-1.Apoptosis. 2011 Jan;16(1):35-44. doi: 10.1007/s10495-010-0538-0. Apoptosis. 2011. PMID: 20812030
-
GRAMD4 mimics p53 and mediates the apoptotic function of p73 at mitochondria.Cell Death Differ. 2011 May;18(5):874-86. doi: 10.1038/cdd.2010.153. Epub 2010 Dec 3. Cell Death Differ. 2011. PMID: 21127500 Free PMC article.
-
Targeting p73--a potential approach in cancer treatment.Curr Pharm Des. 2011;17(6):591-602. doi: 10.2174/138161211795222621. Curr Pharm Des. 2011. PMID: 21391909 Review.
-
The guardians of the genome (p53, TA-p73, and TA-p63) are regulators of tumor suppressor miRNAs network.Cancer Metastasis Rev. 2010 Dec;29(4):613-39. doi: 10.1007/s10555-010-9257-9. Cancer Metastasis Rev. 2010. PMID: 20922462 Review.
Cited by
-
KAT2A/E2F1 Promotes Cell Proliferation and Migration via Upregulating the Expression of UBE2C in Pan-Cancer.Genes (Basel). 2022 Oct 8;13(10):1817. doi: 10.3390/genes13101817. Genes (Basel). 2022. PMID: 36292703 Free PMC article.
-
Identification of ABCA5 among ATP-Binding Cassette Transporter Family as a New Biomarker for Colorectal Cancer.J Oncol. 2022 Jun 22;2022:3399311. doi: 10.1155/2022/3399311. eCollection 2022. J Oncol. 2022. PMID: 35783152 Free PMC article.
-
The broken cycle: E2F dysfunction in cancer.Nat Rev Cancer. 2019 Jun;19(6):326-338. doi: 10.1038/s41568-019-0143-7. Nat Rev Cancer. 2019. PMID: 31053804 Review.
-
A Study on Effect of Oxaliplatin in MicroRNA Expression in Human Colon Cancer.J Cancer. 2018 May 22;9(11):2046-2053. doi: 10.7150/jca.24474. eCollection 2018. J Cancer. 2018. PMID: 29896290 Free PMC article.
-
Data Mining Identifies Six Proteins that Can Act as Prognostic Markers for Head and Neck Squamous Cell Carcinoma.Cell Transplant. 2020 Jan-Dec;29:963689720929308. doi: 10.1177/0963689720929308. Cell Transplant. 2020. PMID: 32452220 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous