

Optic atrophy 1 mediates mitochondria remodeling and dopaminergic neurodegeneration linked to complex I deficiency
- PMID: 22858546
- PMCID: PMC3524632
- DOI: 10.1038/cdd.2012.95
Optic atrophy 1 mediates mitochondria remodeling and dopaminergic neurodegeneration linked to complex I deficiency
Abstract
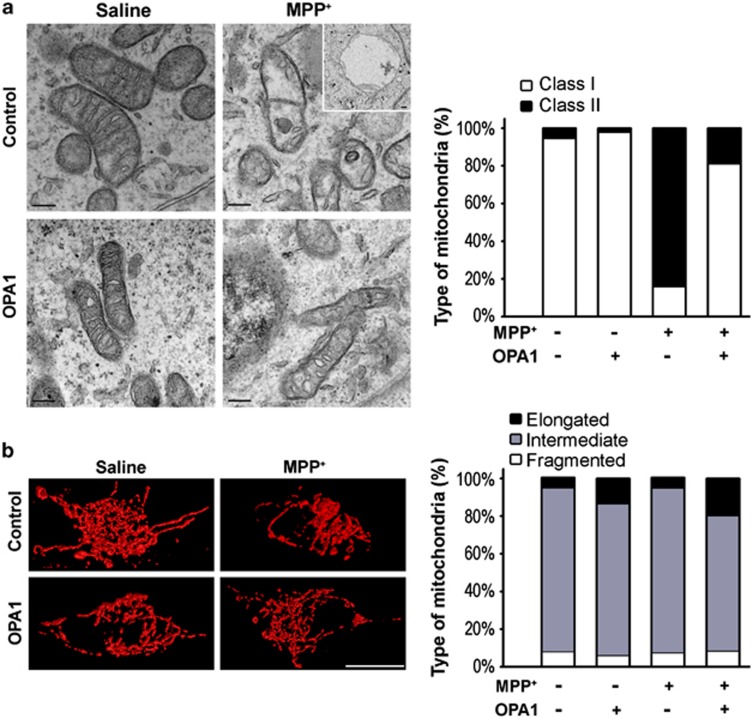
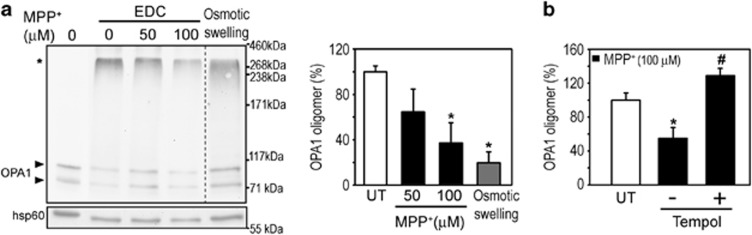
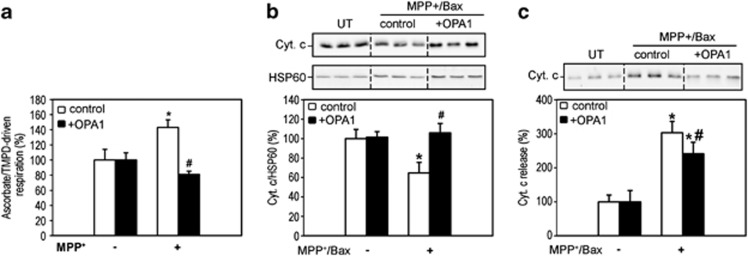
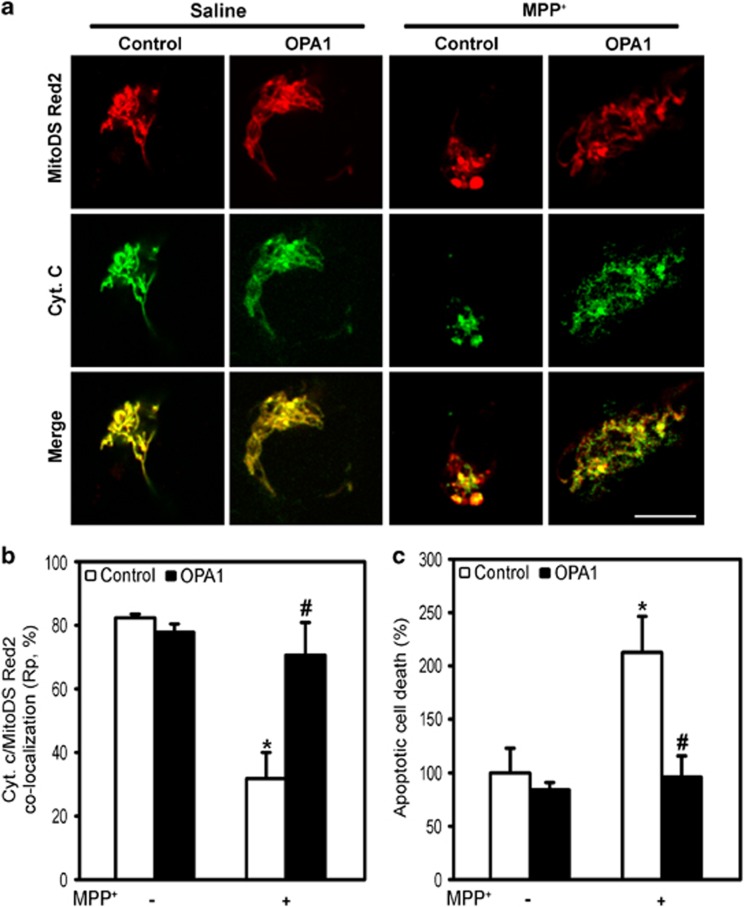
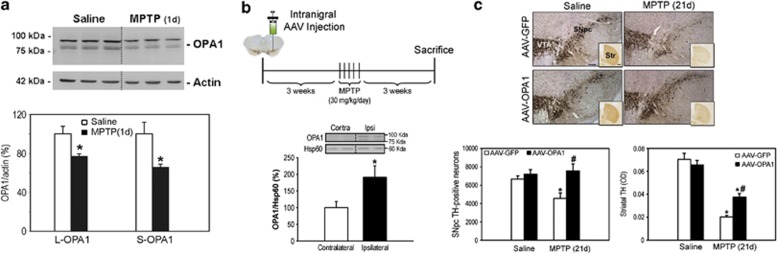
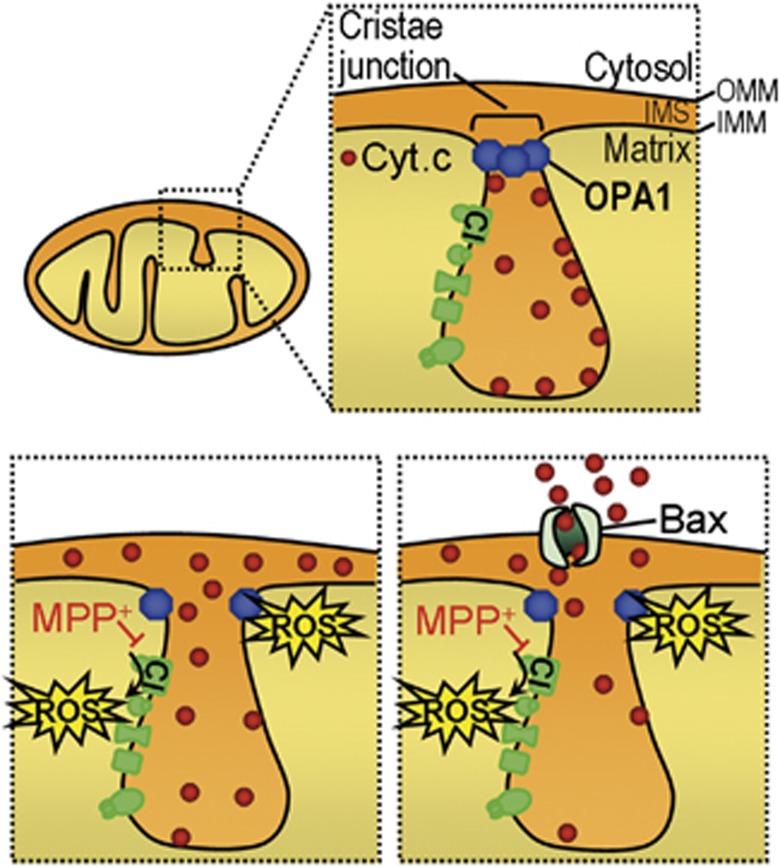
Mitochondrial complex I dysfunction has long been associated with Parkinson's disease (PD). Recent evidence suggests that mitochondrial involvement in PD may extend beyond a sole respiratory deficit and also include perturbations in mitochondrial fusion/fission or ultrastructure. Whether and how alterations in mitochondrial dynamics may relate to the known complex I defects in PD is unclear. Optic atrophy 1 (OPA1), a dynamin-related GTPase of the inner mitochondrial membrane, participates in mitochondrial fusion and apoptotic mitochondrial cristae remodeling. Here we show that complex I inhibition by parkinsonian neurotoxins leads to an oxidative-dependent disruption of OPA1 oligomeric complexes that normally keep mitochondrial cristae junctions tight. As a consequence, affected mitochondria exhibit major structural abnormalities, including cristae disintegration, loss of matrix density and swelling. These changes are not accompanied by mitochondrial fission but a mobilization of cytochrome c from cristae to intermembrane space, thereby lowering the threshold for activation of mitochondria-dependent apoptosis by cell death agonists in compromised neurons. All these pathogenic changes, including mitochondrial structural remodeling and dopaminergic neurodegeneration, are abrogated by OPA1 overexpression, both in vitro and in vivo. Our results identify OPA1 as molecular link between complex I deficiency and alterations in mitochondrial dynamics machinery and point to OPA1 as a novel therapeutic target for complex I cytopathies, such as PD.
Figures
Similar articles
-
Loss of prohibitin membrane scaffolds impairs mitochondrial architecture and leads to tau hyperphosphorylation and neurodegeneration.PLoS Genet. 2012;8(11):e1003021. doi: 10.1371/journal.pgen.1003021. Epub 2012 Nov 8. PLoS Genet. 2012. PMID: 23144624 Free PMC article.
-
OPA1 overexpression ameliorates mitochondrial cristae remodeling, mitochondrial dysfunction, and neuronal apoptosis in prion diseases.Cell Death Dis. 2019 Sep 24;10(10):710. doi: 10.1038/s41419-019-1953-y. Cell Death Dis. 2019. PMID: 31551424 Free PMC article.
-
OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion.Cell. 2006 Jul 14;126(1):177-89. doi: 10.1016/j.cell.2006.06.025. Cell. 2006. PMID: 16839885
-
OPA1: How much do we know to approach therapy?Pharmacol Res. 2018 May;131:199-210. doi: 10.1016/j.phrs.2018.02.018. Epub 2018 Feb 15. Pharmacol Res. 2018. PMID: 29454676 Review.
-
The dynamin GTPase OPA1: more than mitochondria?Biochim Biophys Acta. 2013 Jan;1833(1):176-83. doi: 10.1016/j.bbamcr.2012.08.004. Epub 2012 Aug 11. Biochim Biophys Acta. 2013. PMID: 22902477 Review.
Cited by
-
Mitochondria dynamism: of shape, transport and cell migration.Cell Mol Life Sci. 2014 Jun;71(12):2313-24. doi: 10.1007/s00018-014-1557-8. Epub 2014 Jan 18. Cell Mol Life Sci. 2014. PMID: 24442478 Free PMC article. Review.
-
Fusion or Fission: The Destiny of Mitochondria In Traumatic Brain Injury of Different Severities.Sci Rep. 2017 Aug 23;7(1):9189. doi: 10.1038/s41598-017-09587-2. Sci Rep. 2017. PMID: 28835707 Free PMC article.
-
Mfn2 Overexpression Attenuates MPTP Neurotoxicity In Vivo.Int J Mol Sci. 2021 Jan 9;22(2):601. doi: 10.3390/ijms22020601. Int J Mol Sci. 2021. PMID: 33435331 Free PMC article.
-
Mechanisms and roles of mitophagy in neurodegenerative diseases.CNS Neurosci Ther. 2019 Jul;25(7):859-875. doi: 10.1111/cns.13140. Epub 2019 May 2. CNS Neurosci Ther. 2019. PMID: 31050206 Free PMC article. Review.
-
Too much death can kill you: inhibiting intrinsic apoptosis to treat disease.EMBO J. 2021 Jul 15;40(14):e107341. doi: 10.15252/embj.2020107341. Epub 2021 May 26. EMBO J. 2021. PMID: 34037273 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
