

Cooperative binding of KLF4, pELK-1, and HDAC2 to a G/C repressor element in the SM22α promoter mediates transcriptional silencing during SMC phenotypic switching in vivo
- PMID: 22811558
- PMCID: PMC3517884
- DOI: 10.1161/CIRCRESAHA.112.269811
Cooperative binding of KLF4, pELK-1, and HDAC2 to a G/C repressor element in the SM22α promoter mediates transcriptional silencing during SMC phenotypic switching in vivo
Abstract
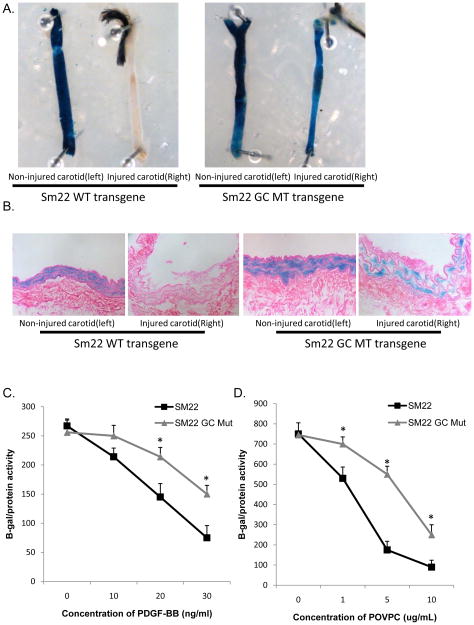
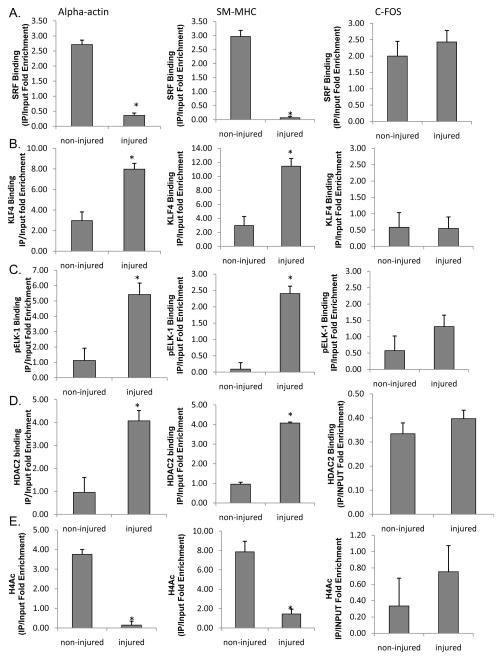
Rationale: We previously identified conserved G/C Repressor elements in the promoters of most smooth muscle cell (SMC) marker genes and demonstrated that mutation of this element within the SM22α promoter nearly abrogated repression of this transgene after vascular wire injury or within lesions of ApoE-/- mice. However, the mechanisms regulating the activity of the G/C Repressor are unknown, although we have previously shown that phenotypic switching of cultured SMC is dependent on Krupple-like factor (KLF)4.
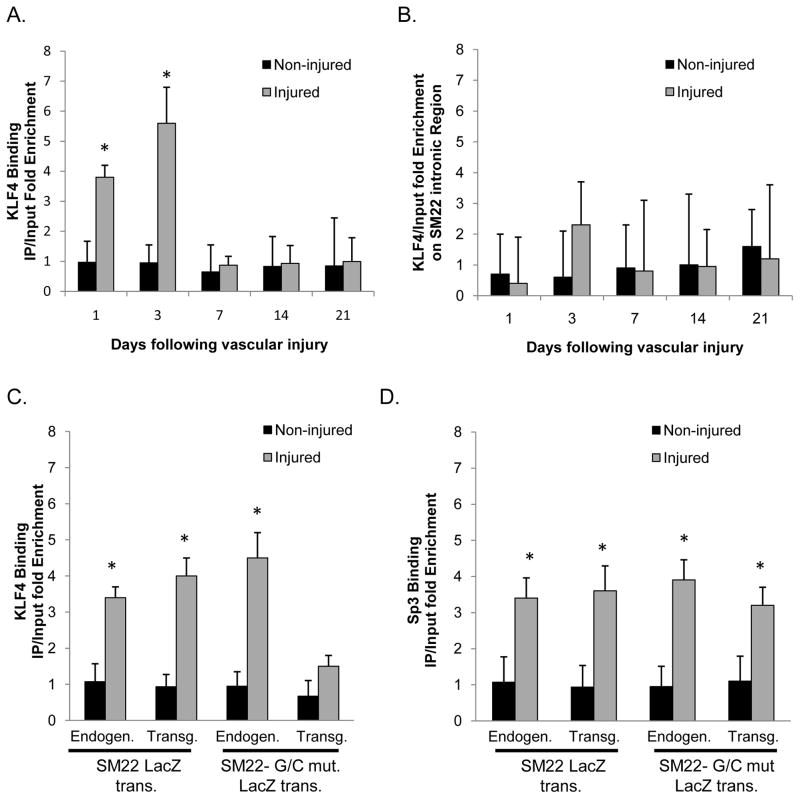
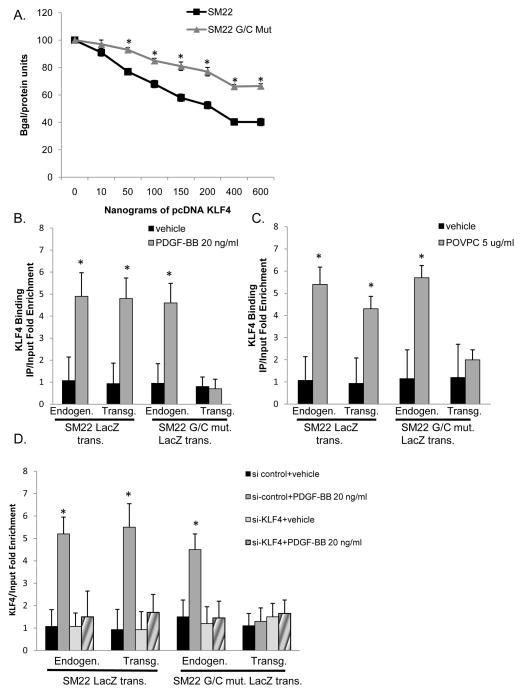
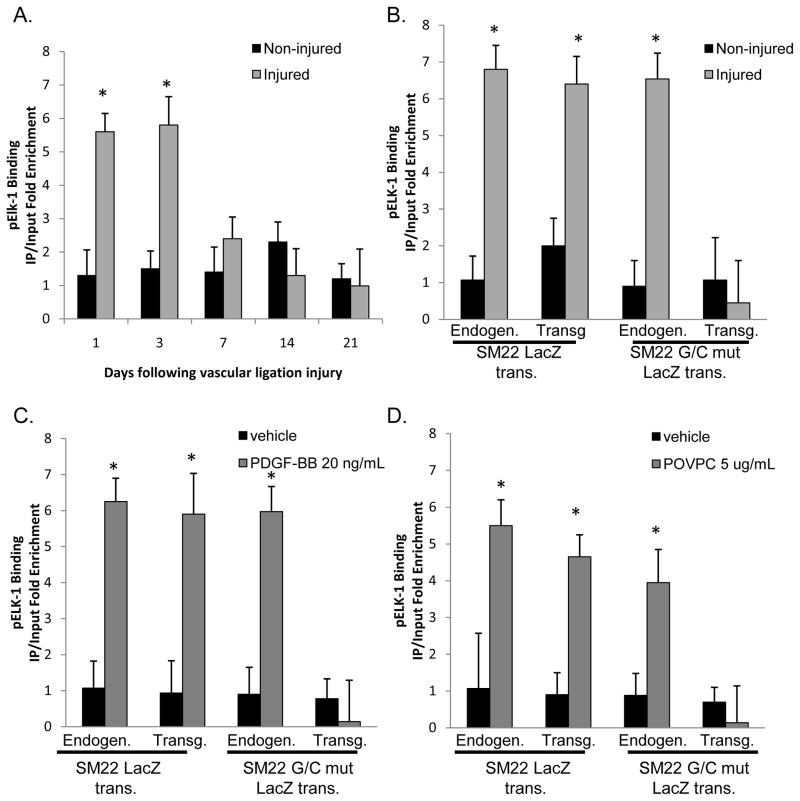
Objective: The goals of the present studies were to ascertain if (1) injury-induced repression of SM22α gene after vascular injury is mediated through KLF4 binding to the G/C Repressor element and (2) the transcriptional repressor activity of KLF4 on SMC marker genes is dependent on cooperative binding with pELK-1 (downstream activator of the mitogen-activated protein kinase pathway) and subsequent recruitment of histone de-acetylase 2 (HDAC2), which mediates epigenetic gene silencing.
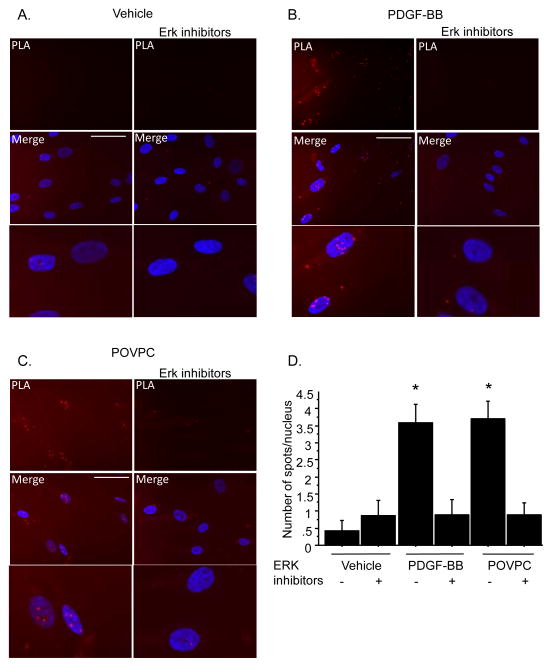
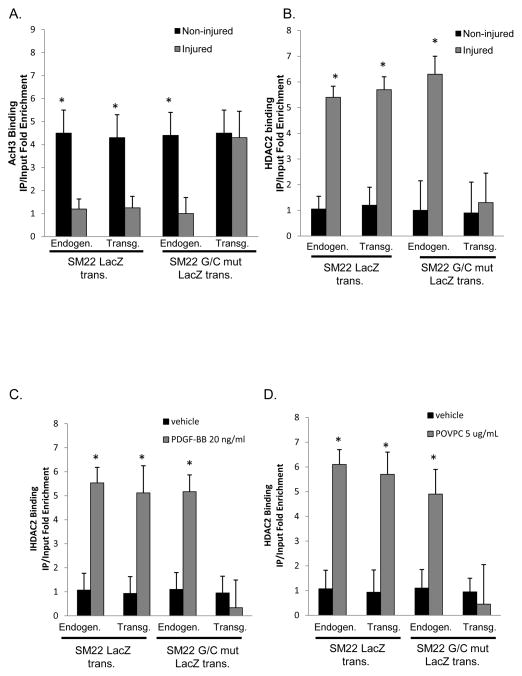
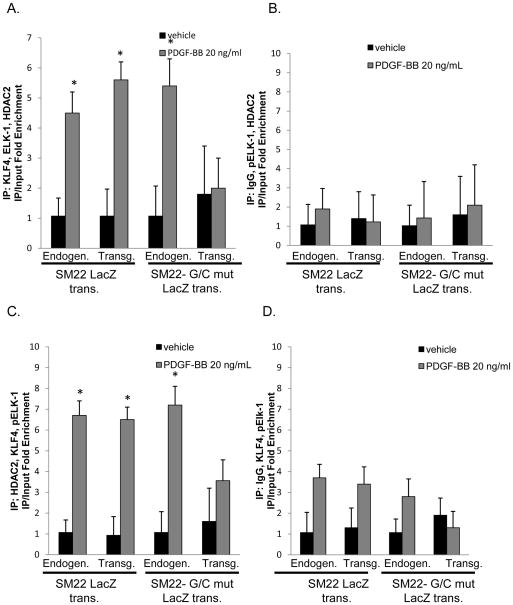
Methods and results: Chromatin immunoprecipitation (ChIP) assays were performed on chromatin derived from carotid arteries of mice having either a wild-type or G/C Repressor mutant SM22α promoter-LacZ transgene. KLF4 and pELK-1 binding to the SM22α promoter was markedly increased after vascular injury and was G/C Repressor dependent. Sequential ChIP assays and proximity ligation analyses in cultured SMC treated with platelet-derived growth factor BB or oxidized phospholipids showed formation of a KLF4, pELK-1, and HDAC2 multiprotein complex dependent on the SM22α G/C Repressor element.
Conclusions: Silencing of SMC marker genes during phenotypic switching is partially mediated by sequential binding of pELK-1 and KLF4 to G/C Repressor elements. The pELK-1-KLF4 complex in turn recruits HDAC2, leading to reduced histone acetylation and epigenetic silencing.
Figures
Comment in
-
Modulation of smooth muscle cell phenotype: the other side of the story.Circ Res. 2012 Aug 31;111(6):659-61. doi: 10.1161/CIRCRESAHA.112.277368. Circ Res. 2012. PMID: 22935528 Free PMC article. No abstract available.
Similar articles
-
A G/C element mediates repression of the SM22alpha promoter within phenotypically modulated smooth muscle cells in experimental atherosclerosis.Circ Res. 2004 Nov 12;95(10):981-8. doi: 10.1161/01.RES.0000147961.09840.fb. Epub 2004 Oct 14. Circ Res. 2004. PMID: 15486317
-
Kruppel-like factor 4, Elk-1, and histone deacetylases cooperatively suppress smooth muscle cell differentiation markers in response to oxidized phospholipids.Am J Physiol Cell Physiol. 2008 Nov;295(5):C1175-82. doi: 10.1152/ajpcell.00288.2008. Epub 2008 Sep 3. Am J Physiol Cell Physiol. 2008. PMID: 18768922 Free PMC article.
-
ATRA activates and PDGF-BB represses the SM22α promoter through KLF4 binding to, or dissociating from, its cis-DNA elements.Cardiovasc Res. 2011 Jun 1;90(3):464-74. doi: 10.1093/cvr/cvr017. Epub 2011 Jan 20. Cardiovasc Res. 2011. PMID: 21252119
-
Multiple repressor pathways contribute to phenotypic switching of vascular smooth muscle cells.Am J Physiol Cell Physiol. 2007 Jan;292(1):C59-69. doi: 10.1152/ajpcell.00394.2006. Epub 2006 Sep 6. Am J Physiol Cell Physiol. 2007. PMID: 16956962 Review.
-
Programming smooth muscle plasticity with chromatin dynamics.Circ Res. 2007 May 25;100(10):1428-41. doi: 10.1161/01.RES.0000266448.30370.a0. Circ Res. 2007. PMID: 17525382 Review.
Cited by
-
Transcription factors: key regulatory targets of vascular smooth muscle cell in atherosclerosis.Mol Med. 2023 Jan 5;29(1):2. doi: 10.1186/s10020-022-00586-2. Mol Med. 2023. PMID: 36604627 Free PMC article. Review.
-
Silencing of microRNA-494 inhibits the neurotoxic Th1 shift via regulating HDAC2-STAT4 cascade in ischaemic stroke.Br J Pharmacol. 2020 Jan;177(1):128-144. doi: 10.1111/bph.14852. Epub 2019 Nov 8. Br J Pharmacol. 2020. PMID: 31465536 Free PMC article.
-
Lysozyme Improves the Inhibitory Effects of Panax notoginseng Saponins on Phenotype Transformation of Vascular Smooth Muscle Cells by Binding to Ginsenoside Re.Front Nutr. 2021 Dec 23;8:795888. doi: 10.3389/fnut.2021.795888. eCollection 2021. Front Nutr. 2021. PMID: 35004822 Free PMC article.
-
ZFP148 (Zinc-Finger Protein 148) Binds Cooperatively With NF-1 (Neurofibromin 1) to Inhibit Smooth Muscle Marker Gene Expression During Abdominal Aortic Aneurysm Formation.Arterioscler Thromb Vasc Biol. 2019 Jan;39(1):73-88. doi: 10.1161/ATVBAHA.118.311136. Arterioscler Thromb Vasc Biol. 2019. PMID: 30580567 Free PMC article.
-
KLF4 regulates abdominal aortic aneurysm morphology and deletion attenuates aneurysm formation.Circulation. 2013 Sep 10;128(11 Suppl 1):S163-74. doi: 10.1161/CIRCULATIONAHA.112.000238. Circulation. 2013. PMID: 24030402 Free PMC article.
References
-
- Chamley-Campbell J, Campbell G, Ross R. The smooth muscle cell in culture. Physiol Reviews. 1979;59:1–6. - PubMed
-
- Owens GK, Kumar MS, Wamhoff BR. Molecular Regulation of Vascular Smooth Muscle Cell Differentiation in Development and Disease. Physiol Rev. 2004;84:767–801. - PubMed
-
- Owens GK. Regulation of Differentiation of Vascular Smooth Muscle Cells. Physiol Rev. 1995;75:487–517. - PubMed
-
- Kawai-Kowase K, Owens GK. Multiple repressor pathways contribute to phenotypic swiching of vascular smooth muscle cells. Am J Physiol Cell Physiol. 2007;292:C59–69. - PubMed
-
- Adam PJ, Regan CJ, Hautmann MB, Owens GK. Positive and negative acting kruppel-like transcription factors bind a transforming growth factor beta control element required for expression of the smooth muscle differentiation marker SM22alpha in vivo. J Biol Chem. 2000;275:37798–806. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
