

Microbial co-occurrence relationships in the human microbiome
- PMID: 22807668
- PMCID: PMC3395616
- DOI: 10.1371/journal.pcbi.1002606
Microbial co-occurrence relationships in the human microbiome
Abstract
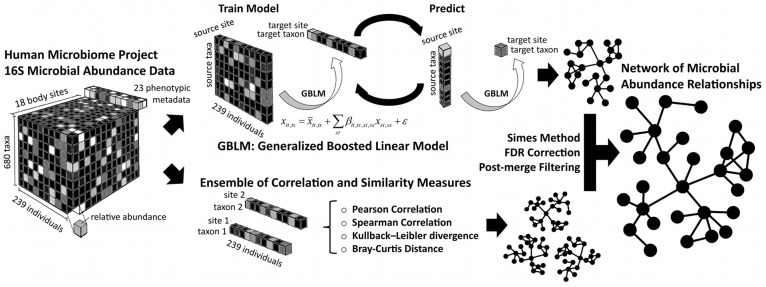
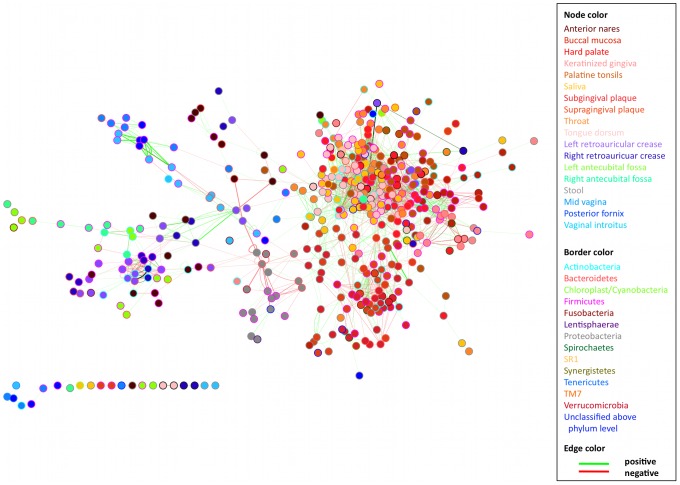
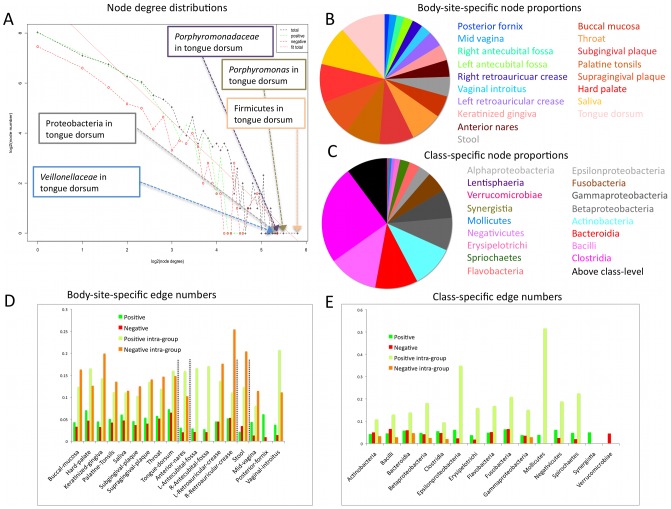
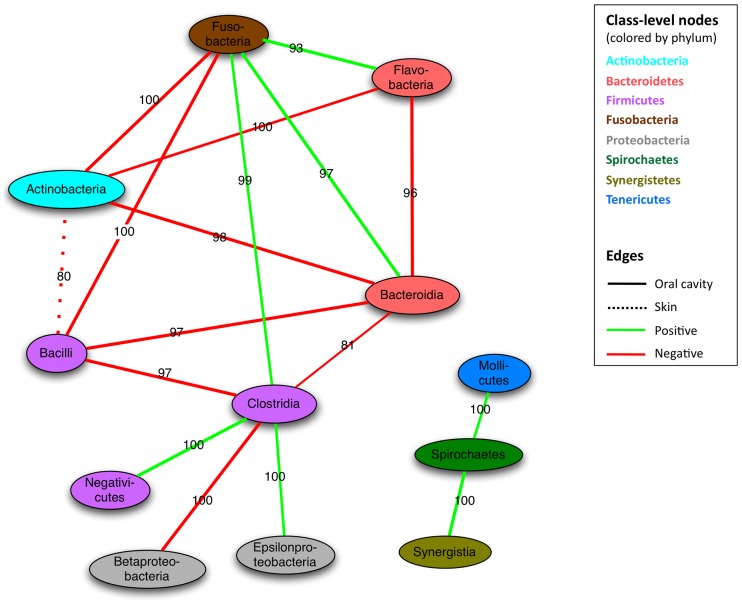
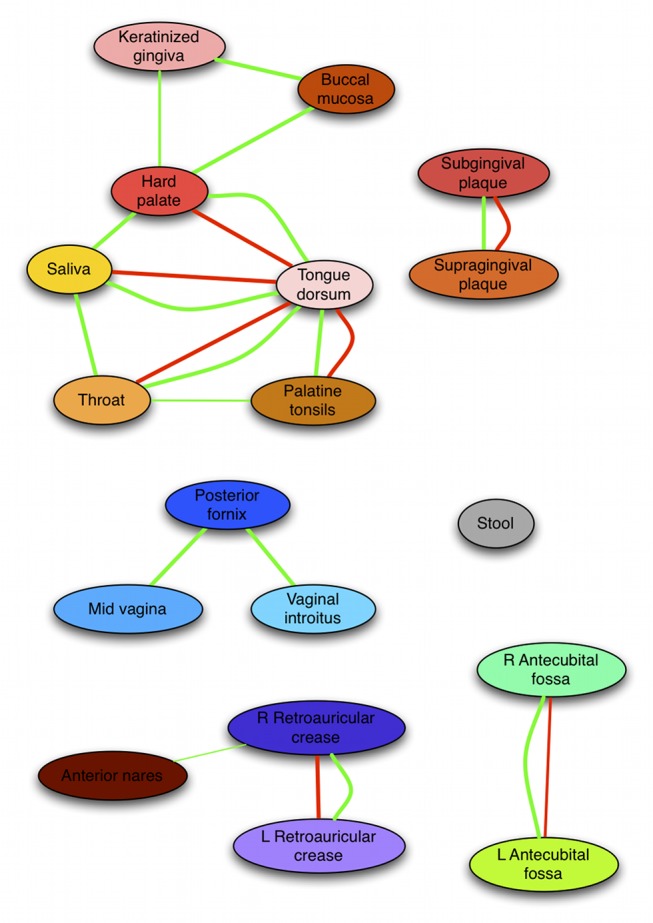
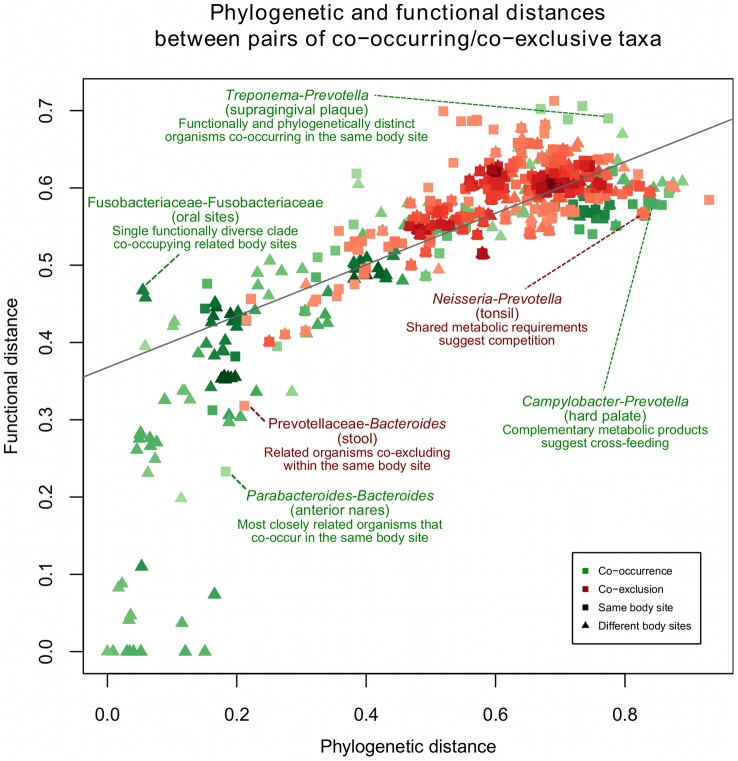
The healthy microbiota show remarkable variability within and among individuals. In addition to external exposures, ecological relationships (both oppositional and symbiotic) between microbial inhabitants are important contributors to this variation. It is thus of interest to assess what relationships might exist among microbes and determine their underlying reasons. The initial Human Microbiome Project (HMP) cohort, comprising 239 individuals and 18 different microbial habitats, provides an unprecedented resource to detect, catalog, and analyze such relationships. Here, we applied an ensemble method based on multiple similarity measures in combination with generalized boosted linear models (GBLMs) to taxonomic marker (16S rRNA gene) profiles of this cohort, resulting in a global network of 3,005 significant co-occurrence and co-exclusion relationships between 197 clades occurring throughout the human microbiome. This network revealed strong niche specialization, with most microbial associations occurring within body sites and a number of accompanying inter-body site relationships. Microbial communities within the oropharynx grouped into three distinct habitats, which themselves showed no direct influence on the composition of the gut microbiota. Conversely, niches such as the vagina demonstrated little to no decomposition into region-specific interactions. Diverse mechanisms underlay individual interactions, with some such as the co-exclusion of Porphyromonaceae family members and Streptococcus in the subgingival plaque supported by known biochemical dependencies. These differences varied among broad phylogenetic groups as well, with the Bacilli and Fusobacteria, for example, both enriched for exclusion of taxa from other clades. Comparing phylogenetic versus functional similarities among bacteria, we show that dominant commensal taxa (such as Prevotellaceae and Bacteroides in the gut) often compete, while potential pathogens (e.g. Treponema and Prevotella in the dental plaque) are more likely to co-occur in complementary niches. This approach thus serves to open new opportunities for future targeted mechanistic studies of the microbial ecology of the human microbiome.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Phylogenetic molecular ecological network of soil microbial communities in response to elevated CO2.mBio. 2011 Jul 26;2(4):e00122-11. doi: 10.1128/mBio.00122-11. Print 2011. mBio. 2011. PMID: 21791581 Free PMC article.
-
Multi-level comparisons of cloacal, skin, feather and nest-associated microbiota suggest considerable influence of horizontal acquisition on the microbiota assembly of sympatric woodlarks and skylarks.Microbiome. 2017 Dec 1;5(1):156. doi: 10.1186/s40168-017-0371-6. Microbiome. 2017. PMID: 29191217 Free PMC article.
-
Ecophylogenetics Clarifies the Evolutionary Association between Mammals and Their Gut Microbiota.mBio. 2018 Sep 11;9(5):e01348-18. doi: 10.1128/mBio.01348-18. mBio. 2018. PMID: 30206171 Free PMC article.
-
Microbial diversity and interactions in subgingival biofilm communities.Front Oral Biol. 2012;15:17-40. doi: 10.1159/000329669. Epub 2011 Nov 11. Front Oral Biol. 2012. PMID: 22142955 Review.
-
Ex vivo systems to study host-microbiota interactions in the gastrointestinal tract.Best Pract Res Clin Gastroenterol. 2013 Feb;27(1):101-13. doi: 10.1016/j.bpg.2013.03.018. Best Pract Res Clin Gastroenterol. 2013. PMID: 23768556 Review.
Cited by
-
Categorization of the gut microbiota: enterotypes or gradients?Nat Rev Microbiol. 2012 Sep;10(9):591-2. doi: 10.1038/nrmicro2859. Nat Rev Microbiol. 2012. PMID: 23066529 No abstract available.
-
Diverse Diets with Consistent Core Microbiome in Wild Bee Pollen Provisions.Insects. 2020 Aug 4;11(8):499. doi: 10.3390/insects11080499. Insects. 2020. PMID: 32759653 Free PMC article.
-
Chapter 12: Human microbiome analysis.PLoS Comput Biol. 2012;8(12):e1002808. doi: 10.1371/journal.pcbi.1002808. Epub 2012 Dec 27. PLoS Comput Biol. 2012. PMID: 23300406 Free PMC article.
-
Biogeography of the ecosystems of the healthy human body.Genome Biol. 2013 Jan 14;14(1):R1. doi: 10.1186/gb-2013-14-1-r1. Genome Biol. 2013. PMID: 23316946 Free PMC article.
-
Exploring the transcriptome of Staphylococcus aureus in its natural niche.Sci Rep. 2016 Sep 19;6:33174. doi: 10.1038/srep33174. Sci Rep. 2016. PMID: 27641137 Free PMC article.
References
-
- Saffo MB. Coming to Terms with a Field - Words and Concepts in Symbiosis (Vol 14, Pg 29, 1993). Symbiosis. 1993;15:181–181.
-
- William Z. Lidicker J. A Clarification of Interactions in Ecological Systems. BioScience. 1979;29:475–477.
-
- Diamond JM. Assembly of species communities. In: Cody ML, Diamond JM, editors. Ecology and Evolution of Communities. Cambridge, MA: Harvard University Press; 1975. pp. 342–444.
-
- Konopka A. What is microbial community ecology? ISME J. 2009;3:1223–1230. - PubMed
-
- Woyke T, Teeling H, Ivanova NN, Huntemann M, Richter M, et al. Symbiotic insights through metagenomic analysis of a microbial consortium. Nature. 2006;443:950–955. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
