

Dominant optic atrophy
- PMID: 22776096
- PMCID: PMC3526509
- DOI: 10.1186/1750-1172-7-46
Dominant optic atrophy
Abstract
DEFINITION OF THE DISEASE: Dominant Optic Atrophy (DOA) is a neuro-ophthalmic condition characterized by a bilateral degeneration of the optic nerves, causing insidious visual loss, typically starting during the first decade of life. The disease affects primary the retinal ganglion cells (RGC) and their axons forming the optic nerve, which transfer the visual information from the photoreceptors to the lateral geniculus in the brain.
Epidemiology: The prevalence of the disease varies from 1/10000 in Denmark due to a founder effect, to 1/30000 in the rest of the world.
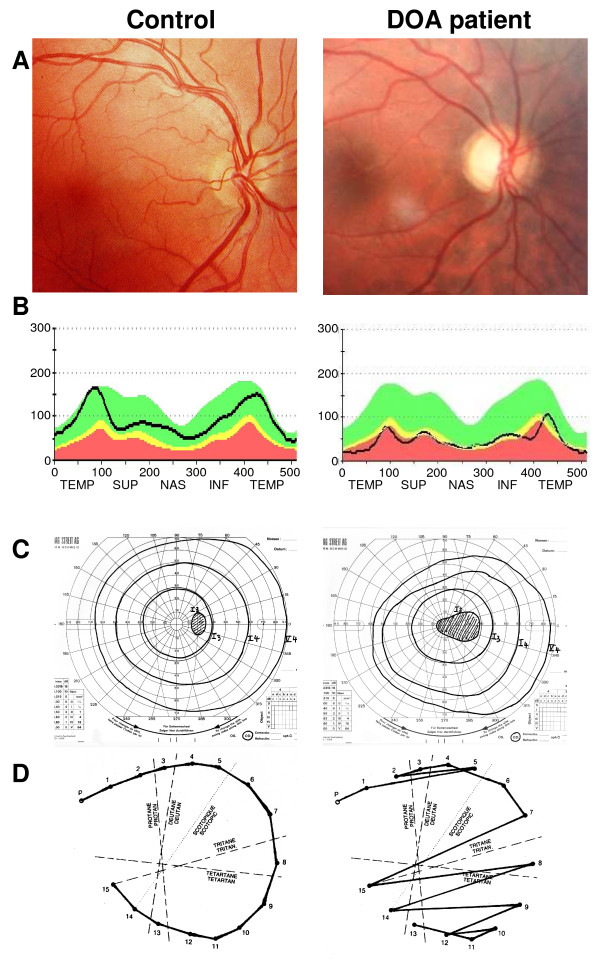
Clinical description: DOA patients usually suffer of moderate visual loss, associated with central or paracentral visual field deficits and color vision defects. The severity of the disease is highly variable, the visual acuity ranging from normal to legal blindness. The ophthalmic examination discloses on fundoscopy isolated optic disc pallor or atrophy, related to the RGC death. About 20% of DOA patients harbour extraocular multi-systemic features, including neurosensory hearing loss, or less commonly chronic progressive external ophthalmoplegia, myopathy, peripheral neuropathy, multiple sclerosis-like illness, spastic paraplegia or cataracts.
Aetiology: Two genes (OPA1, OPA3) encoding inner mitochondrial membrane proteins and three loci (OPA4, OPA5, OPA8) are currently known for DOA. Additional loci and genes (OPA2, OPA6 and OPA7) are responsible for X-linked or recessive optic atrophy. All OPA genes yet identified encode mitochondrial proteins embedded in the inner membrane and ubiquitously expressed, as are the proteins mutated in the Leber Hereditary Optic Neuropathy. OPA1 mutations affect mitochondrial fusion, energy metabolism, control of apoptosis, calcium clearance and maintenance of mitochondrial genome integrity. OPA3 mutations only affect the energy metabolism and the control of apoptosis.
Diagnosis: Patients are usually diagnosed during their early childhood, because of bilateral, mild, otherwise unexplained visual loss related to optic discs pallor or atrophy, and typically occurring in the context of a family history of DOA. Optical Coherence Tomography further discloses non-specific thinning of retinal nerve fiber layer, but a normal morphology of the photoreceptors layers. Abnormal visual evoked potentials and pattern ERG may also reflect the dysfunction of the RGCs and their axons. Molecular diagnosis is provided by the identification of a mutation in the OPA1 gene (75% of DOA patients) or in the OPA3 gene (1% of patients).
Prognosis: Visual loss in DOA may progress during puberty until adulthood, with very slow subsequent chronic progression in most of the cases. On the opposite, in DOA patients with associated extra-ocular features, the visual loss may be more severe over time.
Management: To date, there is no preventative or curative treatment in DOA; severely visually impaired patients may benefit from low vision aids. Genetic counseling is commonly offered and patients are advised to avoid alcohol and tobacco consumption, as well as the use of medications that may interfere with mitochondrial metabolism. Gene and pharmacological therapies for DOA are currently under investigation.
Figures
Similar articles
-
Dominant optic atrophy: Culprit mitochondria in the optic nerve.Prog Retin Eye Res. 2021 Jul;83:100935. doi: 10.1016/j.preteyeres.2020.100935. Epub 2020 Dec 17. Prog Retin Eye Res. 2021. PMID: 33340656 Review.
-
The prevalence and natural history of dominant optic atrophy due to OPA1 mutations.Ophthalmology. 2010 Aug;117(8):1538-46, 1546.e1. doi: 10.1016/j.ophtha.2009.12.038. Epub 2010 Apr 24. Ophthalmology. 2010. PMID: 20417570 Free PMC article.
-
Genetic screening for OPA1 and OPA3 mutations in patients with suspected inherited optic neuropathies.Ophthalmology. 2011 Mar;118(3):558-63. doi: 10.1016/j.ophtha.2010.07.029. Epub 2010 Oct 30. Ophthalmology. 2011. PMID: 21036400 Free PMC article.
-
First cases of dominant optic atrophy in Saudi Arabia: report of two novel OPA1 mutations.J Neuroophthalmol. 2013 Dec;33(4):349-53. doi: 10.1097/WNO.0b013e31829ffb9a. J Neuroophthalmol. 2013. PMID: 24051421
-
Mitochondrial optic neuropathies - disease mechanisms and therapeutic strategies.Prog Retin Eye Res. 2011 Mar;30(2):81-114. doi: 10.1016/j.preteyeres.2010.11.002. Epub 2010 Nov 26. Prog Retin Eye Res. 2011. PMID: 21112411 Free PMC article. Review.
Cited by
-
The human OPA1delTTAG mutation induces adult onset and progressive auditory neuropathy in mice.Cell Mol Life Sci. 2024 Feb 9;81(1):80. doi: 10.1007/s00018-024-05115-4. Cell Mol Life Sci. 2024. PMID: 38334784 Free PMC article.
-
Saccharomyces cerevisiae as a Tool for Studying Mutations in Nuclear Genes Involved in Diseases Caused by Mitochondrial DNA Instability.Genes (Basel). 2021 Nov 24;12(12):1866. doi: 10.3390/genes12121866. Genes (Basel). 2021. PMID: 34946817 Free PMC article. Review.
-
Whole Exome Sequencing Identifies Two Novel Mutations in the Reticulon 4-Interacting Protein 1 Gene in a Chinese Family with Autosomal Recessive Optic Neuropathies.J Mol Neurosci. 2019 Aug;68(4):640-646. doi: 10.1007/s12031-019-01319-7. Epub 2019 May 10. J Mol Neurosci. 2019. PMID: 31077085
-
The Present and Future of Mitochondrial-Based Therapeutics for Eye Disease.Transl Vis Sci Technol. 2021 Jul 1;10(8):4. doi: 10.1167/tvst.10.8.4. Transl Vis Sci Technol. 2021. PMID: 34232272 Free PMC article.
-
Optic neuropathies: the tip of the neurodegeneration iceberg.Hum Mol Genet. 2017 Oct 1;26(R2):R139-R150. doi: 10.1093/hmg/ddx273. Hum Mol Genet. 2017. PMID: 28977448 Free PMC article. Review.
References
-
- Kjer P. Infantile optic atrophy with dominant mode of inheritance: a clinical and genetic study of 19 Danish families. Acta Ophthalmol Suppl. 1959;164(Supp 54):1–147. - PubMed
-
- Thiselton DL, Alexander C, Taanman JW, Brooks S, Rosenberg T, Eiberg H, Andreasson S, Van Regemorter N, Munier FL, Moore AT. et al.A comprehensive survey of mutations in the OPA1 gene in patients with autosomal dominant optic atrophy. Invest Ophthalmol Vis Sci. 2002;43(6):1715–1724. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
