

Mechanisms of fibrosis: therapeutic translation for fibrotic disease
- PMID: 22772564
- PMCID: PMC3405917
- DOI: 10.1038/nm.2807
Mechanisms of fibrosis: therapeutic translation for fibrotic disease
Abstract
Fibrosis is a pathological feature of most chronic inflammatory diseases. Fibrosis, or scarring, is defined by the accumulation of excess extracellular matrix components. If highly progressive, the fibrotic process eventually leads to organ malfunction and death. Fibrosis affects nearly every tissue in the body. Here we discuss how key components of the innate and adaptive immune response contribute to the pathogenesis of fibrosis. We also describe how cell-intrinsic changes in important structural cells can perpetuate the fibrotic response by regulating the differentiation, recruitment, proliferation and activation of extracellular matrix-producing myofibroblasts. Finally, we highlight some of the key mechanisms and pathways of fibrosis that are being targeted as potential therapies for a variety of important human diseases.
Figures


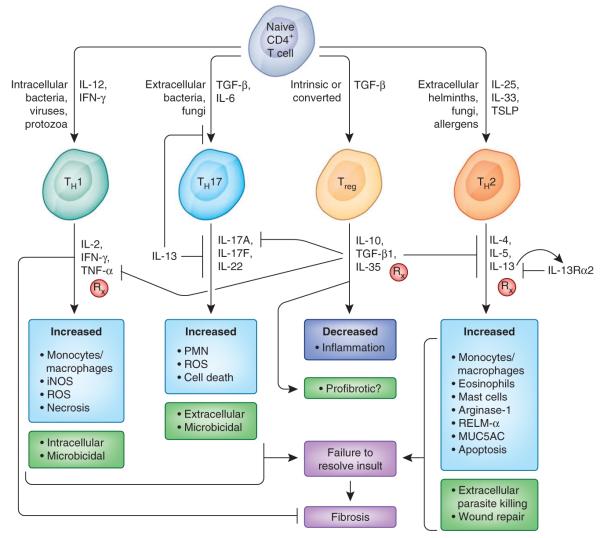

Similar articles
-
The Roles of Immune Cells in the Pathogenesis of Fibrosis.Int J Mol Sci. 2020 Jul 22;21(15):5203. doi: 10.3390/ijms21155203. Int J Mol Sci. 2020. PMID: 32708044 Free PMC article. Review.
-
Fibrosis: Types, Effects, Markers, Mechanisms for Disease Progression, and Its Relation with Oxidative Stress, Immunity, and Inflammation.Int J Mol Sci. 2023 Feb 16;24(4):4004. doi: 10.3390/ijms24044004. Int J Mol Sci. 2023. PMID: 36835428 Free PMC article. Review.
-
Human Fibrotic Diseases: Current Challenges in Fibrosis Research.Methods Mol Biol. 2017;1627:1-23. doi: 10.1007/978-1-4939-7113-8_1. Methods Mol Biol. 2017. PMID: 28836191 Review.
-
Regulation of fibrosis by the immune system.Adv Immunol. 2006;89:245-88. doi: 10.1016/S0065-2776(05)89006-6. Adv Immunol. 2006. PMID: 16682276 Review.
-
Organ and tissue fibrosis: Molecular signals, cellular mechanisms and translational implications.Mol Aspects Med. 2019 Feb;65:2-15. doi: 10.1016/j.mam.2018.06.003. Epub 2018 Jun 30. Mol Aspects Med. 2019. PMID: 29958900 Review.
Cited by
-
PPARs: modulating lipotoxicity and thus inhibiting fibrosis.Hormones (Athens). 2024 Nov 6. doi: 10.1007/s42000-024-00612-4. Online ahead of print. Hormones (Athens). 2024. PMID: 39500811 Review.
-
Fibrosis-related biomarkers and large and small vessel disease: the Cardiovascular Health Study.Atherosclerosis. 2015 Apr;239(2):539-46. doi: 10.1016/j.atherosclerosis.2015.02.020. Epub 2015 Feb 16. Atherosclerosis. 2015. PMID: 25725316 Free PMC article.
-
Tocotrienol-Rich Fractions Offer Potential to Suppress Pulmonary Fibrosis Progression.Int J Mol Sci. 2022 Nov 18;23(22):14331. doi: 10.3390/ijms232214331. Int J Mol Sci. 2022. PMID: 36430808 Free PMC article.
-
Advances in cellular and molecular pathways of salivary gland damage in Sjögren's syndrome.Front Immunol. 2024 Jul 10;15:1405126. doi: 10.3389/fimmu.2024.1405126. eCollection 2024. Front Immunol. 2024. PMID: 39050857 Free PMC article. Review.
-
ER exit in physiology and disease.Front Mol Biosci. 2024 Jan 18;11:1352970. doi: 10.3389/fmolb.2024.1352970. eCollection 2024. Front Mol Biosci. 2024. PMID: 38314136 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
