

Xenome--a tool for classifying reads from xenograft samples
- PMID: 22689758
- PMCID: PMC3371868
- DOI: 10.1093/bioinformatics/bts236
Xenome--a tool for classifying reads from xenograft samples
Abstract
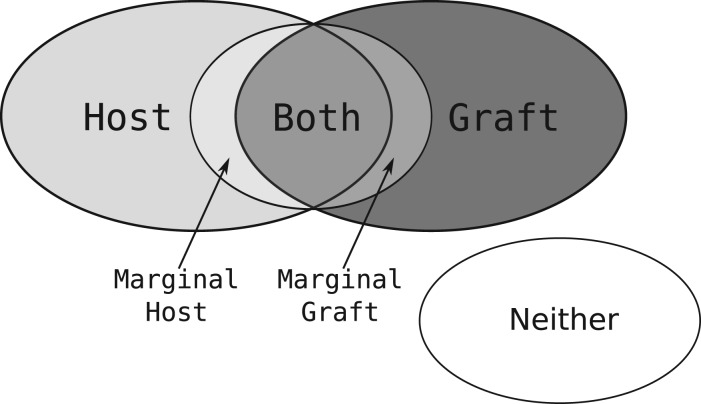
Motivation: Shotgun sequence read data derived from xenograft material contains a mixture of reads arising from the host and reads arising from the graft. Classifying the read mixture to separate the two allows for more precise analysis to be performed.
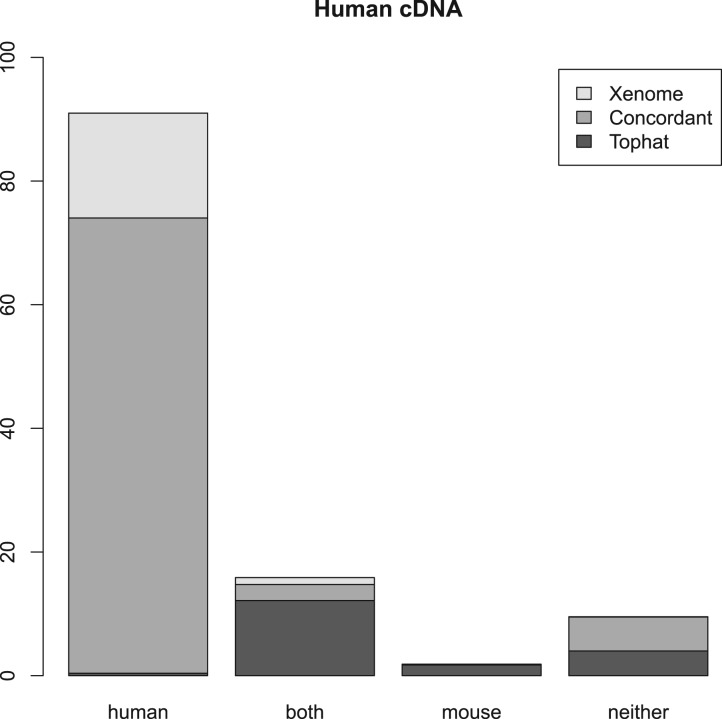
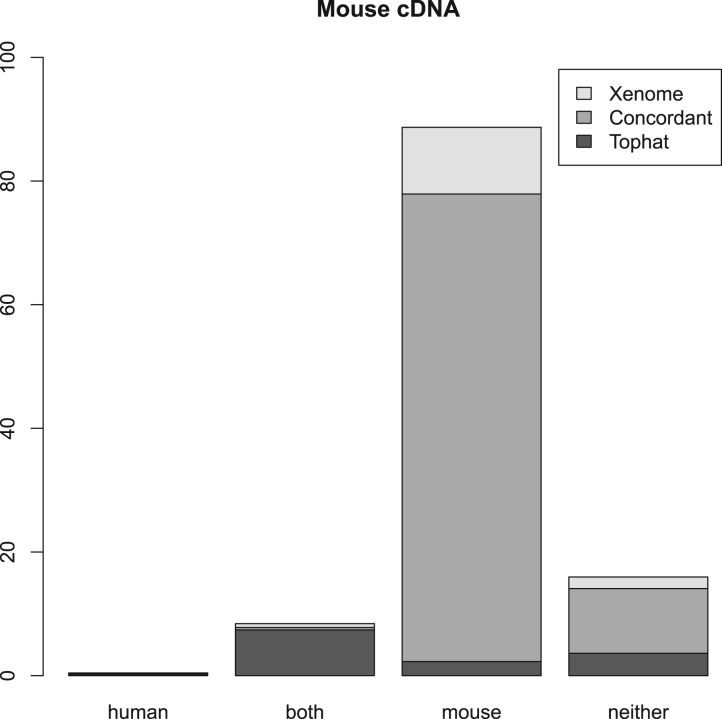
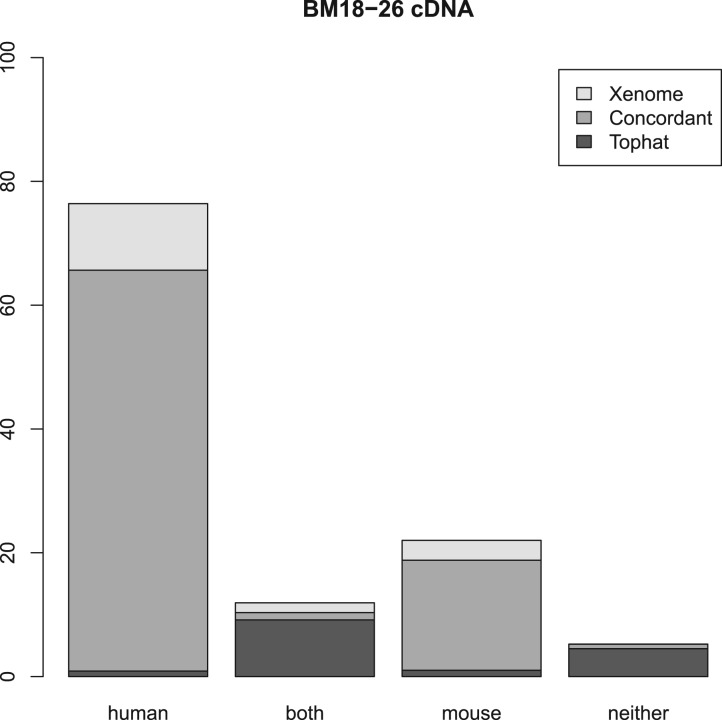
Results: We present a technique, with an associated tool Xenome, which performs fast, accurate and specific classification of xenograft-derived sequence read data. We have evaluated it on RNA-Seq data from human, mouse and human-in-mouse xenograft datasets.
Availability: Xenome is available for non-commercial use from http://www.nicta.com.au/bioinformatics.
Figures
Similar articles
-
OSA: a fast and accurate alignment tool for RNA-Seq.Bioinformatics. 2012 Jul 15;28(14):1933-4. doi: 10.1093/bioinformatics/bts294. Epub 2012 May 15. Bioinformatics. 2012. PMID: 22592379
-
Hierarchical analysis of RNA-seq reads improves the accuracy of allele-specific expression.Bioinformatics. 2018 Jul 1;34(13):2177-2184. doi: 10.1093/bioinformatics/bty078. Bioinformatics. 2018. PMID: 29444201 Free PMC article.
-
RNA-Seq gene expression estimation with read mapping uncertainty.Bioinformatics. 2010 Feb 15;26(4):493-500. doi: 10.1093/bioinformatics/btp692. Epub 2009 Dec 18. Bioinformatics. 2010. PMID: 20022975 Free PMC article.
-
FBB: a fast Bayesian-bound tool to calibrate RNA-seq aligners.Bioinformatics. 2017 Jan 15;33(2):210-218. doi: 10.1093/bioinformatics/btw608. Epub 2016 Sep 23. Bioinformatics. 2017. PMID: 27663496
-
A comparison of next-generation sequencing analysis methods for cancer xenograft samples.J Genet Genomics. 2018 Jul 20;45(7):345-350. doi: 10.1016/j.jgg.2018.07.001. Epub 2018 Jul 25. J Genet Genomics. 2018. PMID: 30055875 Review.
Cited by
-
Increased predominance of the matured ventricular subtype in embryonic stem cell-derived cardiomyocytes in vivo.Sci Rep. 2020 Jul 17;10(1):11883. doi: 10.1038/s41598-020-68373-9. Sci Rep. 2020. PMID: 32681032 Free PMC article.
-
Estrogen receptor positive breast cancers have patient specific hormone sensitivities and rely on progesterone receptor.Nat Commun. 2022 Jun 6;13(1):3127. doi: 10.1038/s41467-022-30898-0. Nat Commun. 2022. PMID: 35668111 Free PMC article.
-
A Therapeutic Strategy for Chemotherapy-Resistant Gastric Cancer via Destabilization of Both β-Catenin and RAS.Cancers (Basel). 2019 Apr 8;11(4):496. doi: 10.3390/cancers11040496. Cancers (Basel). 2019. PMID: 30965636 Free PMC article.
-
Colorectal Cancer Organoid-Stroma Biobank Allows Subtype-Specific Assessment of Individualized Therapy Responses.Cancer Discov. 2023 Oct 5;13(10):2192-2211. doi: 10.1158/2159-8290.CD-23-0050. Cancer Discov. 2023. PMID: 37489084 Free PMC article.
-
Antitumor efficacy of XPO1 inhibitor Selinexor in KRAS-mutant lung adenocarcinoma patient-derived xenografts.Transl Oncol. 2021 Oct;14(10):101179. doi: 10.1016/j.tranon.2021.101179. Epub 2021 Jul 17. Transl Oncol. 2021. PMID: 34284202 Free PMC article.
References
-
- Arbitman Y., et al. 2010 IEEE 51st Annual Symposium on Foundations of Computer Science. Los Alamos California: IEEE Computer Society; 2010. Backyard cuckoo hashing: Constant worst-case operations with a succinct representation; pp. 787–796.
-
- Conway T.C., Bromage A.J. Succinct data structures for assembling large genomes. Bioinformatics. 2011;27:479–486. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
