

A novel mutation in LEPRE1 that eliminates only the KDEL ER- retrieval sequence causes non-lethal osteogenesis imperfecta
- PMID: 22615817
- PMCID: PMC3352923
- DOI: 10.1371/journal.pone.0036809
A novel mutation in LEPRE1 that eliminates only the KDEL ER- retrieval sequence causes non-lethal osteogenesis imperfecta
Abstract
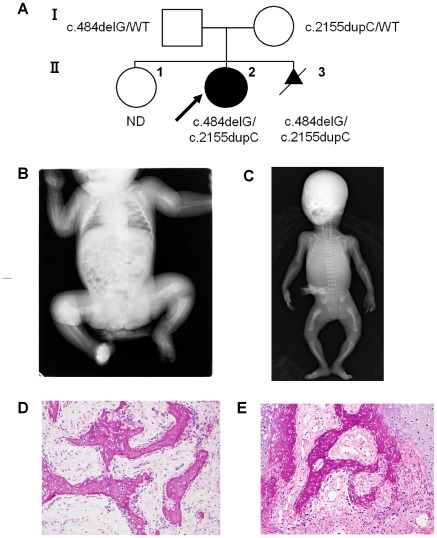
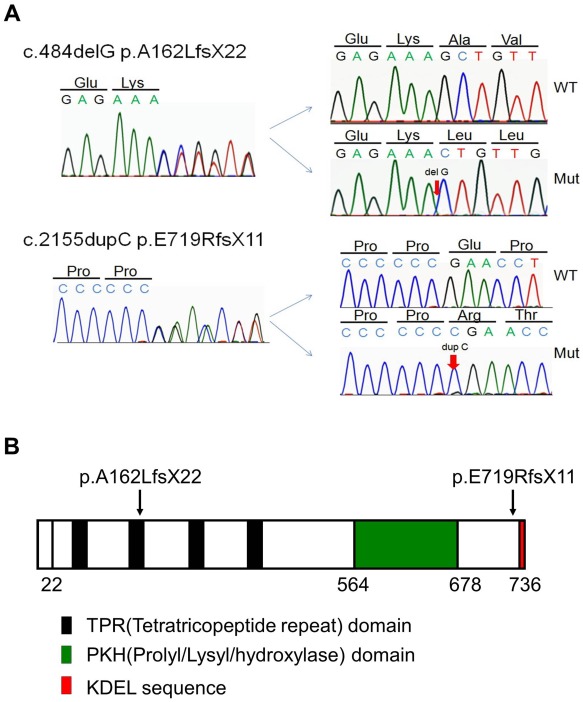
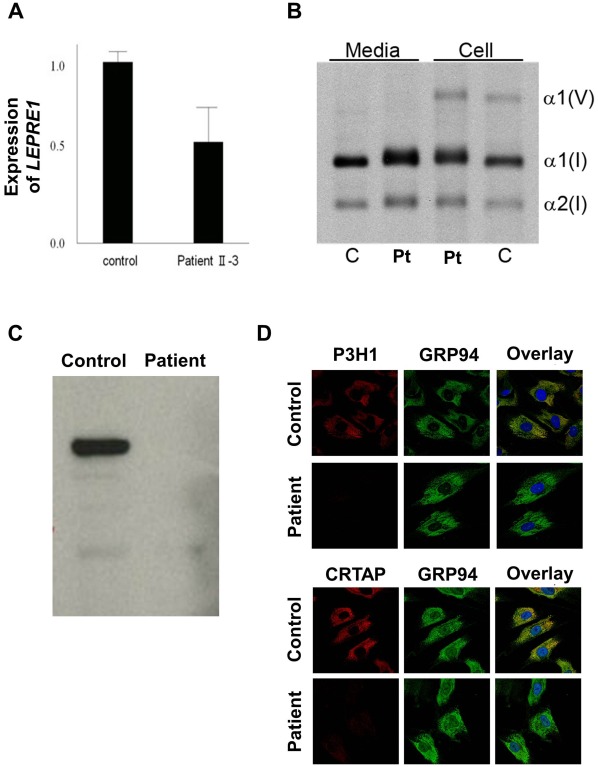
Prolyl 3-hydroxylase 1 (P3H1), encoded by the LEPRE1 gene, forms a molecular complex with cartilage-associated protein (CRTAP) and cyclophilin B (encoded by PPIB) in the endoplasmic reticulum (ER). This complex is responsible for one step in collagen post-translational modification, the prolyl 3-hydroxylation of specific proline residues, specifically α1(I) Pro986. P3H1 provides the enzymatic activity of the complex and has a Lys-Asp-Glu-Leu (KDEL) ER-retrieval sequence at the carboxyl terminus. Loss of function mutations in LEPRE1 lead to the Pro986 residue remaining unmodified and lead to slow folding and excessive helical post-translational modification of type I collagen, which is seen in both dominant and recessive osteogenesis imperfecta (OI). Here, we present the case of siblings with non-lethal OI due to novel compound heterozygous mutations in LEPRE1 (c.484delG and c.2155dupC). The results of RNA analysis and real-time PCR suggest that mRNA with c.2155dupC escapes from nonsense-mediated RNA decay. Without the KDEL ER- retrieval sequence, the product of the c.2155dupC variant cannot be retained in the ER. This is the first report of a mutation in LEPRE1 that eliminates only the KDEL ER-retrieval sequence, whereas other functional domains remain intact. Our study shows, for the first time, that the KDEL ER- retrieval sequence is essential for P3H1 functionality and that a defect in KDEL is sufficient for disease onset.
Conflict of interest statement
Figures
Similar articles
-
Recessive osteogenesis imperfecta caused by LEPRE1 mutations: clinical documentation and identification of the splice form responsible for prolyl 3-hydroxylation.J Med Genet. 2009 Apr;46(4):233-41. doi: 10.1136/jmg.2008.062729. Epub 2008 Dec 16. J Med Genet. 2009. PMID: 19088120
-
Prolyl 3-hydroxylase 1 and CRTAP are mutually stabilizing in the endoplasmic reticulum collagen prolyl 3-hydroxylation complex.Hum Mol Genet. 2010 Jan 15;19(2):223-34. doi: 10.1093/hmg/ddp481. Epub 2009 Oct 21. Hum Mol Genet. 2010. PMID: 19846465 Free PMC article.
-
Null mutations in LEPRE1 and CRTAP cause severe recessive osteogenesis imperfecta.Cell Tissue Res. 2010 Jan;339(1):59-70. doi: 10.1007/s00441-009-0872-0. Epub 2009 Oct 28. Cell Tissue Res. 2010. PMID: 19862557 Free PMC article. Review.
-
Mutations in PPIB (cyclophilin B) delay type I procollagen chain association and result in perinatal lethal to moderate osteogenesis imperfecta phenotypes.Hum Mol Genet. 2011 Apr 15;20(8):1595-609. doi: 10.1093/hmg/ddr037. Epub 2011 Jan 31. Hum Mol Genet. 2011. PMID: 21282188 Free PMC article.
-
Components of the collagen prolyl 3-hydroxylation complex are crucial for normal bone development.Cell Cycle. 2007 Jul 15;6(14):1675-81. doi: 10.4161/cc.6.14.4474. Epub 2007 May 18. Cell Cycle. 2007. PMID: 17630507 Review.
Cited by
-
Collagen transport and related pathways in Osteogenesis Imperfecta.Hum Genet. 2021 Aug;140(8):1121-1141. doi: 10.1007/s00439-021-02302-2. Epub 2021 Jun 24. Hum Genet. 2021. PMID: 34169326 Free PMC article. Review.
-
Milder presentation of osteogenesis imperfecta type VIII due to compound heterozygosity for a predicted loss-of-function variant and novel missense variant in P3H1-further expansion of the phenotypic spectrum.Cold Spring Harb Mol Case Stud. 2023 Mar 24;9(1):a006260. doi: 10.1101/mcs.a006260. Print 2023 Feb. Cold Spring Harb Mol Case Stud. 2023. PMID: 36963805 Free PMC article.
-
An additional function of the rough endoplasmic reticulum protein complex prolyl 3-hydroxylase 1·cartilage-associated protein·cyclophilin B: the CXXXC motif reveals disulfide isomerase activity in vitro.J Biol Chem. 2013 Nov 1;288(44):31437-46. doi: 10.1074/jbc.M113.498063. Epub 2013 Sep 16. J Biol Chem. 2013. PMID: 24043621 Free PMC article.
-
Differential effects of collagen prolyl 3-hydroxylation on skeletal tissues.PLoS Genet. 2014 Jan;10(1):e1004121. doi: 10.1371/journal.pgen.1004121. Epub 2014 Jan 23. PLoS Genet. 2014. PMID: 24465224 Free PMC article.
-
Cytoskeleton and nuclear lamina affection in recessive osteogenesis imperfecta: A functional proteomics perspective.J Proteomics. 2017 Sep 7;167:46-59. doi: 10.1016/j.jprot.2017.08.007. Epub 2017 Aug 9. J Proteomics. 2017. PMID: 28802583 Free PMC article.
References
-
- Körkkö J, Ala-Kokko L, De Paepe A, Nuytinck L, Earley J, et al. Analysis of the COL1A1 and COL1A2 genes by PCR amplification and scanning by conformation-sensitive gel electrophoresis identifies only COL1A1 mutations in 15 patients with osteogenesis imperfecta type I: identification of common sequences of null-allele mutations. Am J Hum Genet. 1998;62:98–110. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
