

Peptide identification by tandem mass spectrometry with alternate fragmentation modes
- PMID: 22595789
- PMCID: PMC3434779
- DOI: 10.1074/mcp.R112.018556
Peptide identification by tandem mass spectrometry with alternate fragmentation modes
Abstract
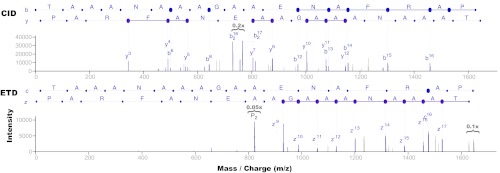
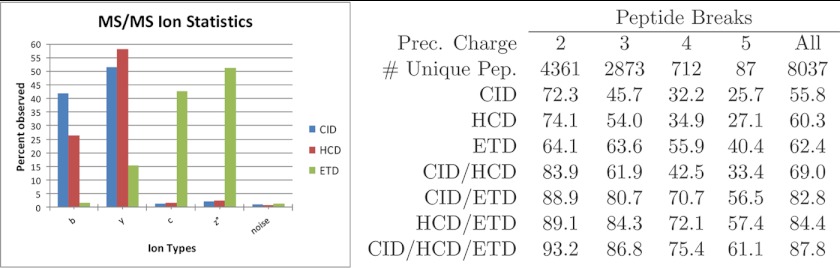
The high-throughput nature of proteomics mass spectrometry is enabled by a productive combination of data acquisition protocols and the computational tools used to interpret the resulting spectra. One of the key components in mainstream protocols is the generation of tandem mass (MS/MS) spectra by peptide fragmentation using collision induced dissociation, the approach currently used in the large majority of proteomics experiments to routinely identify hundreds to thousands of proteins from single mass spectrometry runs. Complementary to these, alternative peptide fragmentation methods such as electron capture/transfer dissociation and higher-energy collision dissociation have consistently achieved significant improvements in the identification of certain classes of peptides, proteins, and post-translational modifications. Recognizing these advantages, mass spectrometry instruments now conveniently support fine-tuned methods that automatically alternate between peptide fragmentation modes for either different types of peptides or for acquisition of multiple MS/MS spectra from each peptide. But although these developments have the potential to substantially improve peptide identification, their routine application requires corresponding adjustments to the software tools and procedures used for automated downstream processing. This review discusses the computational implications of alternative and alternate modes of MS/MS peptide fragmentation and addresses some practical aspects of using such protocols for identification of peptides and post-translational modifications.
Figures
Similar articles
-
Improving the identification rate of endogenous peptides using electron transfer dissociation and collision-induced dissociation.J Proteome Res. 2013 Dec 6;12(12):5410-21. doi: 10.1021/pr400446z. Epub 2013 Oct 3. J Proteome Res. 2013. PMID: 24032530
-
[Recent advances in glycopeptide enrichment and mass spectrometry data interpretation approaches for glycoproteomics analyses].Se Pu. 2021 Oct;39(10):1045-1054. doi: 10.3724/SP.J.1123.2021.06011. Se Pu. 2021. PMID: 34505426 Free PMC article. Review. Chinese.
-
ETISEQ--an algorithm for automated elution time ion sequencing of concurrently fragmented peptides for mass spectrometry-based proteomics.BMC Bioinformatics. 2009 Aug 10;10:244. doi: 10.1186/1471-2105-10-244. BMC Bioinformatics. 2009. PMID: 19664259 Free PMC article.
-
The spectral networks paradigm in high throughput mass spectrometry.Mol Biosyst. 2012 Oct;8(10):2535-44. doi: 10.1039/c2mb25085c. Mol Biosyst. 2012. PMID: 22610447 Free PMC article. Review.
-
Interpretation of collision-induced fragmentation tandem mass spectra of posttranslationally modified peptides.Methods Mol Biol. 2007;367:169-94. doi: 10.1385/1-59745-275-0:169. Methods Mol Biol. 2007. PMID: 17185776
Cited by
-
De Novo MS/MS Sequencing of Native Human Antibodies.J Proteome Res. 2017 Jan 6;16(1):45-54. doi: 10.1021/acs.jproteome.6b00608. Epub 2016 Nov 2. J Proteome Res. 2017. PMID: 27779884 Free PMC article.
-
Current algorithmic solutions for peptide-based proteomics data generation and identification.Curr Opin Biotechnol. 2013 Feb;24(1):31-8. doi: 10.1016/j.copbio.2012.10.013. Epub 2012 Nov 8. Curr Opin Biotechnol. 2013. PMID: 23142544 Free PMC article. Review.
-
Accurate Prediction of y Ions in Beam-Type Collision-Induced Dissociation Using Deep Learning.Anal Chem. 2022 Jun 7;94(22):7752-7758. doi: 10.1021/acs.analchem.1c03184. Epub 2022 May 24. Anal Chem. 2022. PMID: 35609248 Free PMC article.
-
Human Long Noncoding RNA Interactome: Detection, Characterization and Function.Int J Mol Sci. 2020 Feb 4;21(3):1027. doi: 10.3390/ijms21031027. Int J Mol Sci. 2020. PMID: 32033158 Free PMC article. Review.
-
The generating function approach for Peptide identification in spectral networks.J Comput Biol. 2015 May;22(5):353-66. doi: 10.1089/cmb.2014.0165. Epub 2014 Nov 25. J Comput Biol. 2015. PMID: 25423621 Free PMC article.
References
-
- Nilsson T., Mann M., Aebersold R., Yates J. R., 3rd, Bairoch A., Bergeron J. J. (2010) Mass spectrometry in high-throughput proteomics: ready for the big time. Nat. Methods 7, 681–685 - PubMed
-
- Larsen M. R., Trelle M. B., Thingholm T. E., Jensen O. N. (2006) Analysis of posttranslational modifications of proteins by tandem mass spectrometry. Bio. Technique, 40, 790–798 - PubMed
-
- Domon B., Aebersold R. (2010) Options and considerations when selecting a quantitative proteomics strategy. Nat. Biotechnol. 28, 710–721 - PubMed
-
- Eng J. K., Searle B. C., Clauser K. R., Tabb D. L. (2011) A face in the crowd: recognizing peptides through database search. Mol. Cell. Proteomics 10, 10.1074/mcp.R111.009522 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
