

Lithium reduces BACE1 overexpression, β amyloid accumulation, and spatial learning deficits in mice with traumatic brain injury
- PMID: 22583494
- PMCID: PMC3430485
- DOI: 10.1089/neu.2012.2449
Lithium reduces BACE1 overexpression, β amyloid accumulation, and spatial learning deficits in mice with traumatic brain injury
Abstract
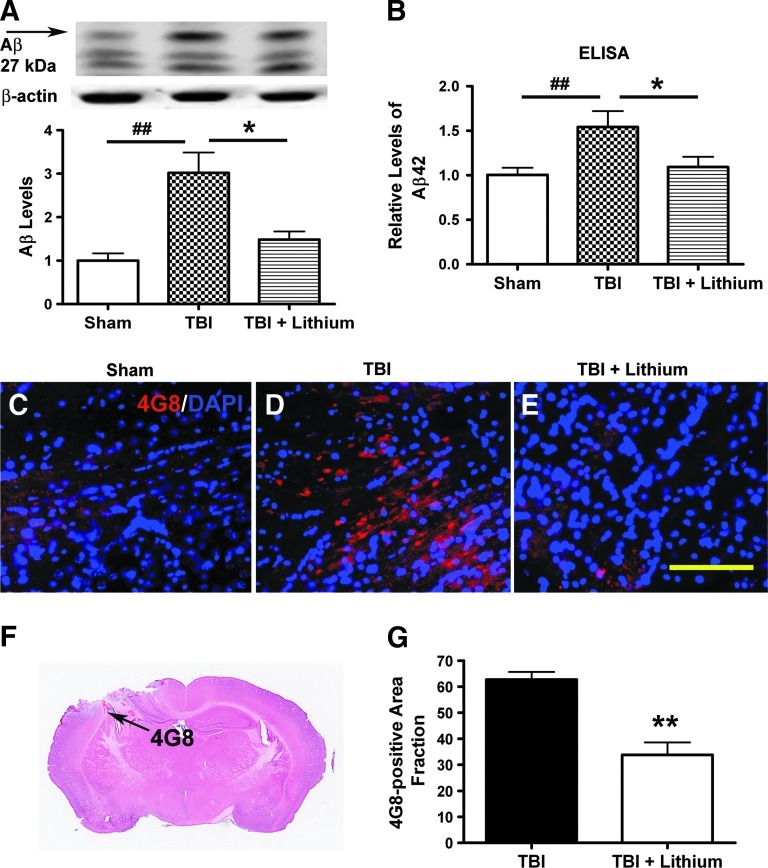
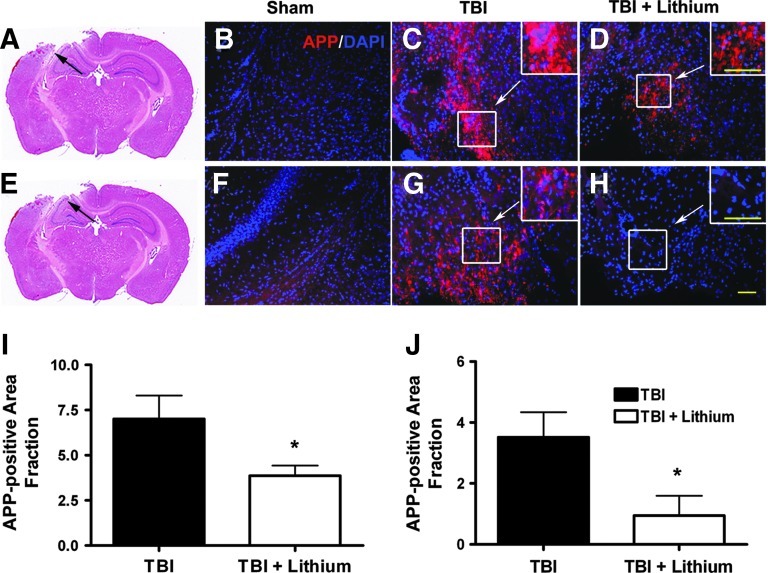
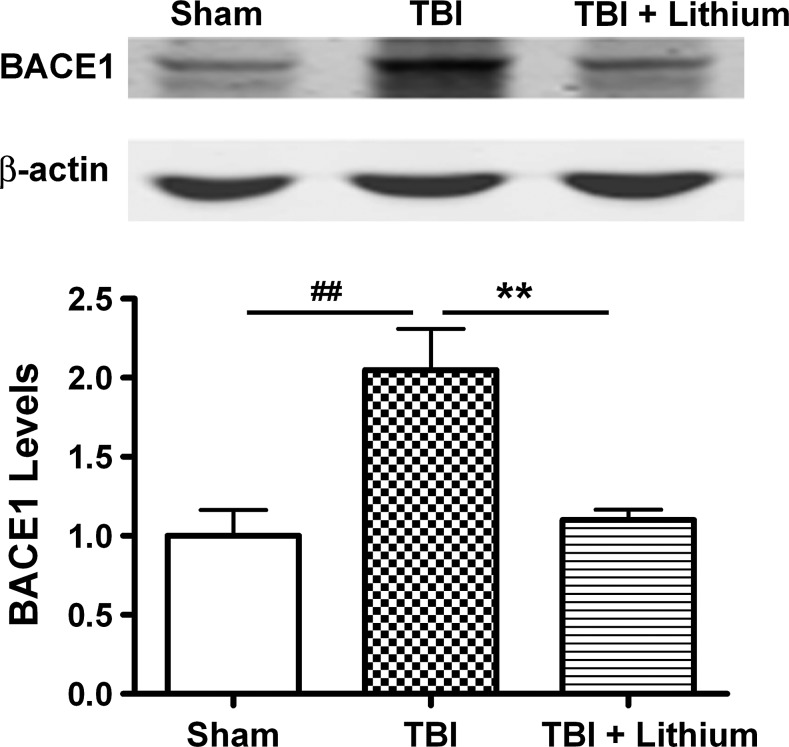
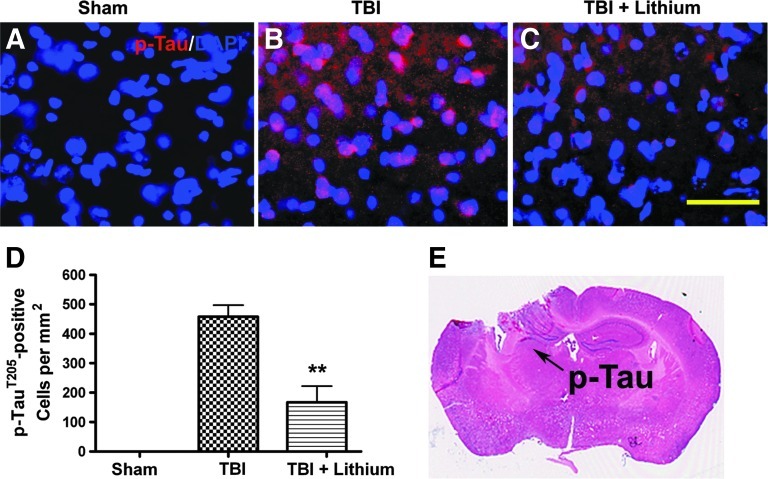
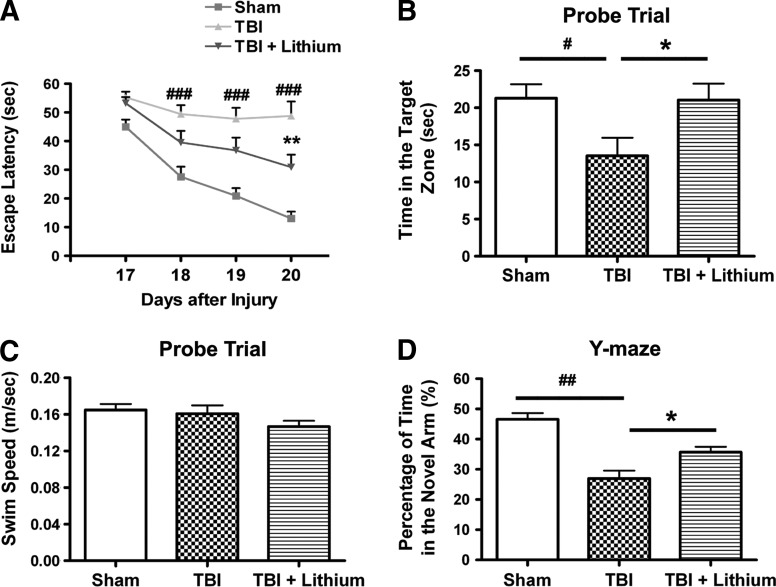
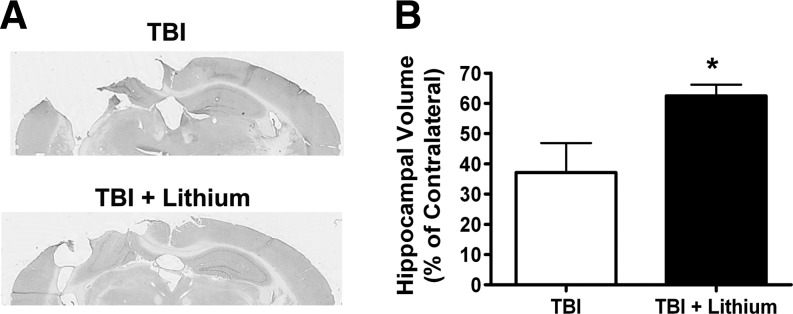
Traumatic brain injury (TBI) leads to both acute injury and long-term neurodegeneration, and is a major risk factor for developing Alzheimer's disease (AD). Beta amyloid (Aβ) peptide deposits in the brain are one of the pathological hallmarks of AD. Aβ levels increase after TBI in animal models and in patients with head trauma, and reducing Aβ levels after TBI has beneficial effects. Lithium is known to be neuroprotective in various models of neurodegenerative disease, and can reduce Aβ generation by modulating glycogen synthase kinase-3 (GSK-3) activity. In this study we explored whether lithium would reduce Aβ load after TBI, and improve learning and memory in a mouse TBI model. Lithium chloride (1.5 mEq/kg, IP) was administered 15 min after TBI, and once daily thereafter for up to 3 weeks. At 3 days after injury, lithium attenuated TBI-induced Aβ load increases, amyloid precursor protein (APP) accumulation, and β-APP-cleaving enzyme-1 (BACE1) overexpression in the corpus callosum and hippocampus. Increased Tau protein phosphorylation in the thalamus was also attenuated after lithium treatment following TBI at the same time point. Notably, lithium treatment significantly improved spatial learning and memory in the Y-maze test conducted 10 days after TBI, and in the Morris water maze test performed 17-20 days post-TBI, in association with increased hippocampal preservation. Thus post-insult treatment with lithium appears to alleviate the TBI-induced Aβ load and consequently improves spatial memory. Our findings suggest that lithium is a potentially useful agent for managing memory impairments after TBI or other head trauma.
Figures
Similar articles
-
Detrimental effect of genetic inhibition of B-site APP-cleaving enzyme 1 on functional outcome after controlled cortical impact in young adult mice.J Neurotrauma. 2011 Sep;28(9):1855-61. doi: 10.1089/neu.2011.1759. Epub 2011 Aug 29. J Neurotrauma. 2011. PMID: 21639727 Free PMC article.
-
Berberine Alleviates Amyloid-Beta Pathology in the Brain of APP/PS1 Transgenic Mice via Inhibiting β/γ-Secretases Activity and Enhancing α-Secretases.Curr Alzheimer Res. 2018;15(11):1045-1052. doi: 10.2174/1567205015666180702105740. Curr Alzheimer Res. 2018. PMID: 29962345
-
Long-term oral melatonin alleviates memory deficits, reduces amyloid-β deposition associated with downregulation of BACE1 and mitophagy in APP/PS1 transgenic mice.Neurosci Lett. 2020 Sep 14;735:135192. doi: 10.1016/j.neulet.2020.135192. Epub 2020 Jun 30. Neurosci Lett. 2020. PMID: 32619650
-
A new avenue for lithium: intervention in traumatic brain injury.ACS Chem Neurosci. 2014 Jun 18;5(6):422-33. doi: 10.1021/cn500040g. Epub 2014 Apr 11. ACS Chem Neurosci. 2014. PMID: 24697257 Free PMC article. Review.
-
Glycogen synthase kinase-3 inhibitors: Rescuers of cognitive impairments.Pharmacol Ther. 2014 Jan;141(1):1-12. doi: 10.1016/j.pharmthera.2013.07.010. Epub 2013 Jul 31. Pharmacol Ther. 2014. PMID: 23916593 Free PMC article. Review.
Cited by
-
Posttrauma cotreatment with lithium and valproate: reduction of lesion volume, attenuation of blood-brain barrier disruption, and improvement in motor coordination in mice with traumatic brain injury.J Neurosurg. 2013 Sep;119(3):766-73. doi: 10.3171/2013.6.JNS13135. Epub 2013 Jul 12. J Neurosurg. 2013. PMID: 23848820 Free PMC article.
-
Performance of Male and Female C57BL/6J Mice on Motor and Cognitive Tasks Commonly Used in Pre-Clinical Traumatic Brain Injury Research.J Neurotrauma. 2016 May 1;33(9):880-94. doi: 10.1089/neu.2015.3977. Epub 2015 Aug 12. J Neurotrauma. 2016. PMID: 25951234 Free PMC article.
-
Mechanical Stress as the Common Denominator between Chronic Inflammation, Cancer, and Alzheimer's Disease.Front Oncol. 2015 Sep 17;5:197. doi: 10.3389/fonc.2015.00197. eCollection 2015. Front Oncol. 2015. PMID: 26442209 Free PMC article. Review.
-
Acute blast injury reduces brain abeta in two rodent species.Front Neurol. 2012 Dec 21;3:177. doi: 10.3389/fneur.2012.00177. eCollection 2012. Front Neurol. 2012. PMID: 23267342 Free PMC article.
-
Mild traumatic brain injury induces microvascular injury and accelerates Alzheimer-like pathogenesis in mice.Acta Neuropathol Commun. 2021 Apr 23;9(1):74. doi: 10.1186/s40478-021-01178-7. Acta Neuropathol Commun. 2021. PMID: 33892818 Free PMC article.
References
-
- Abrahamson E.E. Ikonomovic M.D. Dixon C.E. DeKosky S.T. Simvastatin therapy prevents brain trauma-induced increases in beta-amyloid peptide levels. Ann. Neurol. 2009;66:407–414. - PubMed
-
- Blasko I. Beer R. Bigl M. Apelt J. Franz G. Rudzki D. Ransmayr G. Kampfl A. Schliebs R. Experimental traumatic brain injury in rats stimulates the expression, production and activity of Alzheimer's disease beta-secretase (BACE-1) J. Neural Transm. 2004;111:523–536. - PubMed
-
- Bramlett H.M. Kraydieh S. Green E.J. Dietrich W.D. Temporal and regional patterns of axonal damage following traumatic brain injury: a beta-amyloid precursor protein immunocytochemical study in rats. J. Neuropathol. Exp. Neurol. 1997;56:1132–1141. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
