

Sequential application of anticancer drugs enhances cell death by rewiring apoptotic signaling networks
- PMID: 22579283
- PMCID: PMC3501264
- DOI: 10.1016/j.cell.2012.03.031
Sequential application of anticancer drugs enhances cell death by rewiring apoptotic signaling networks
Abstract
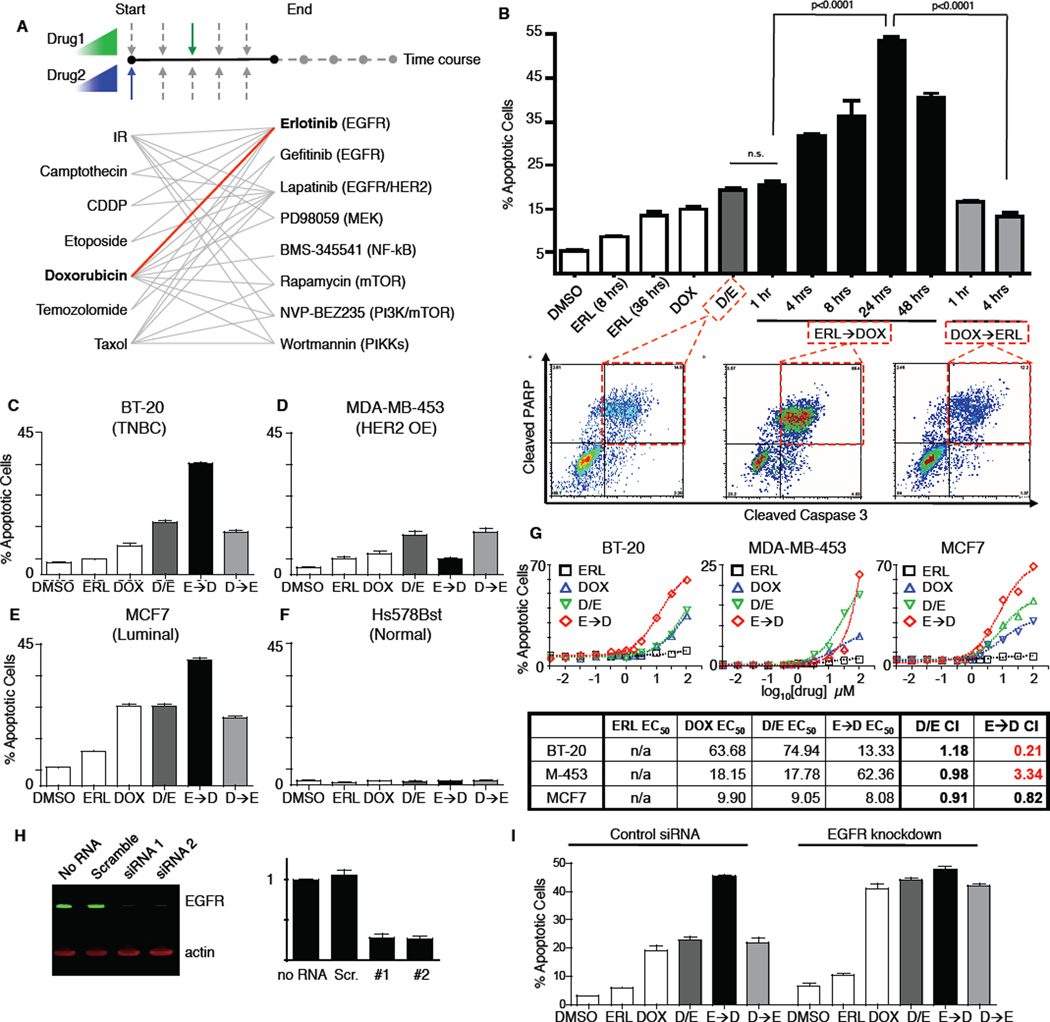
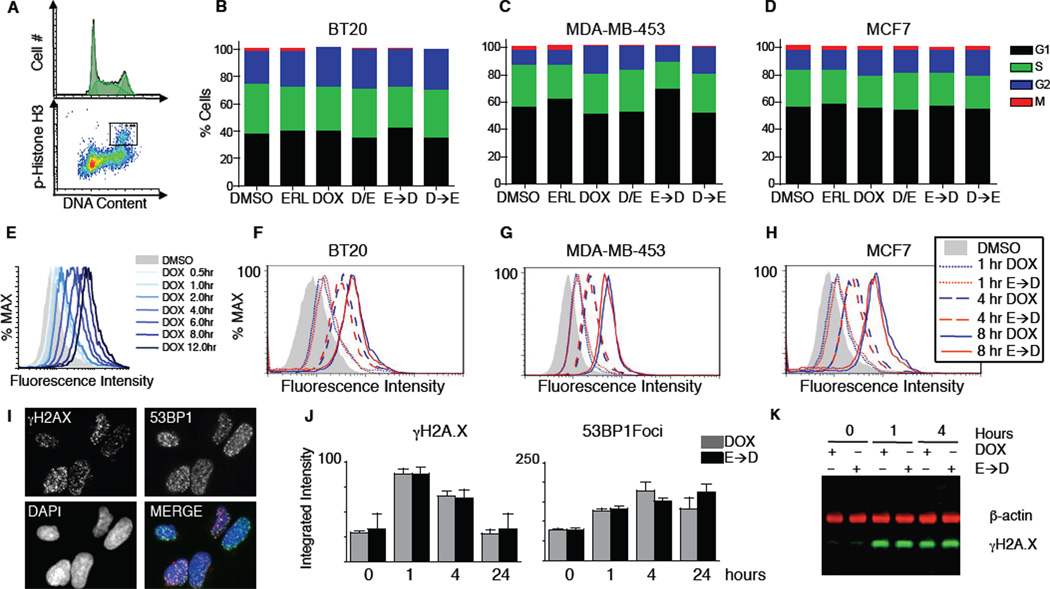
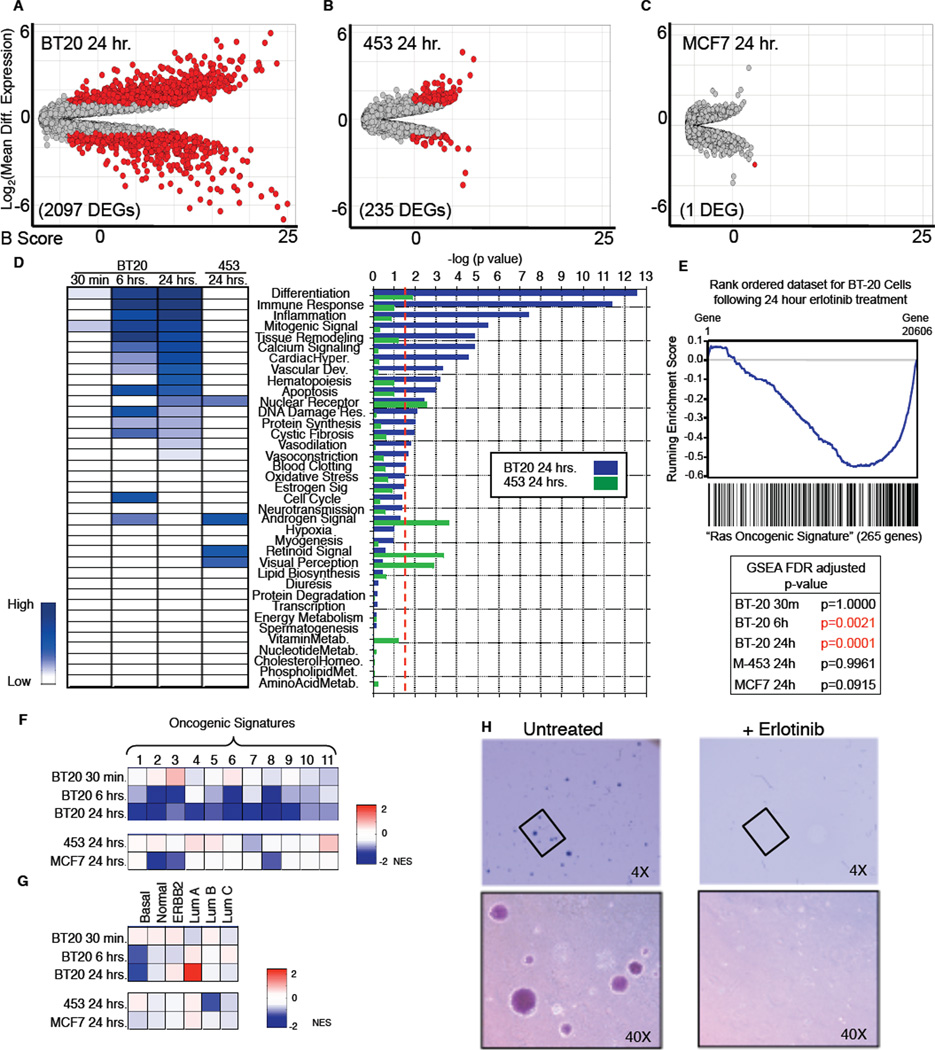
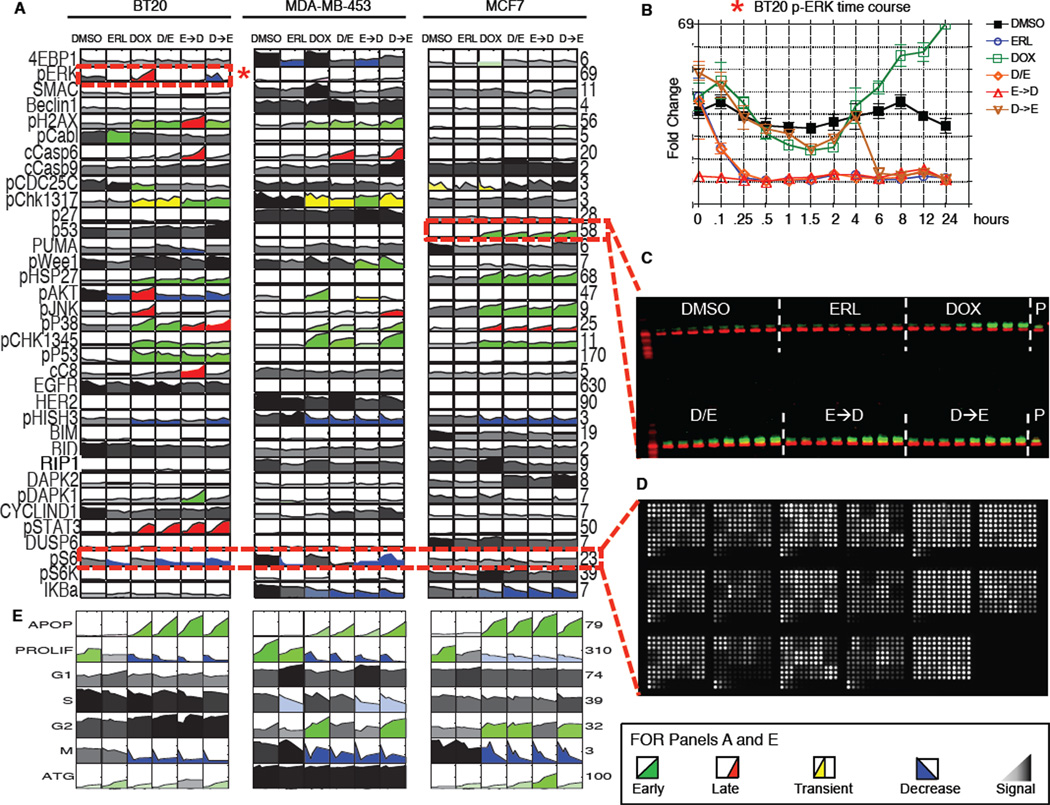
Crosstalk and complexity within signaling pathways and their perturbation by oncogenes limit component-by-component approaches to understanding human disease. Network analysis of how normal and oncogenic signaling can be rewired by drugs may provide opportunities to target tumors with high specificity and efficacy. Using targeted inhibition of oncogenic signaling pathways, combined with DNA-damaging chemotherapy, we report that time-staggered EGFR inhibition, but not simultaneous coadministration, dramatically sensitizes a subset of triple-negative breast cancer cells to genotoxic drugs. Systems-level analysis-using high-density time-dependent measurements of signaling networks, gene expression profiles, and cell phenotypic responses in combination with mathematical modeling-revealed an approach for altering the intrinsic state of the cell through dynamic rewiring of oncogenic signaling pathways. This process converts these cells to a less tumorigenic state that is more susceptible to DNA damage-induced cell death by reactivation of an extrinsic apoptotic pathway whose function is suppressed in the oncogene-addicted state.
Copyright © 2012 Elsevier Inc. All rights reserved.
Figures
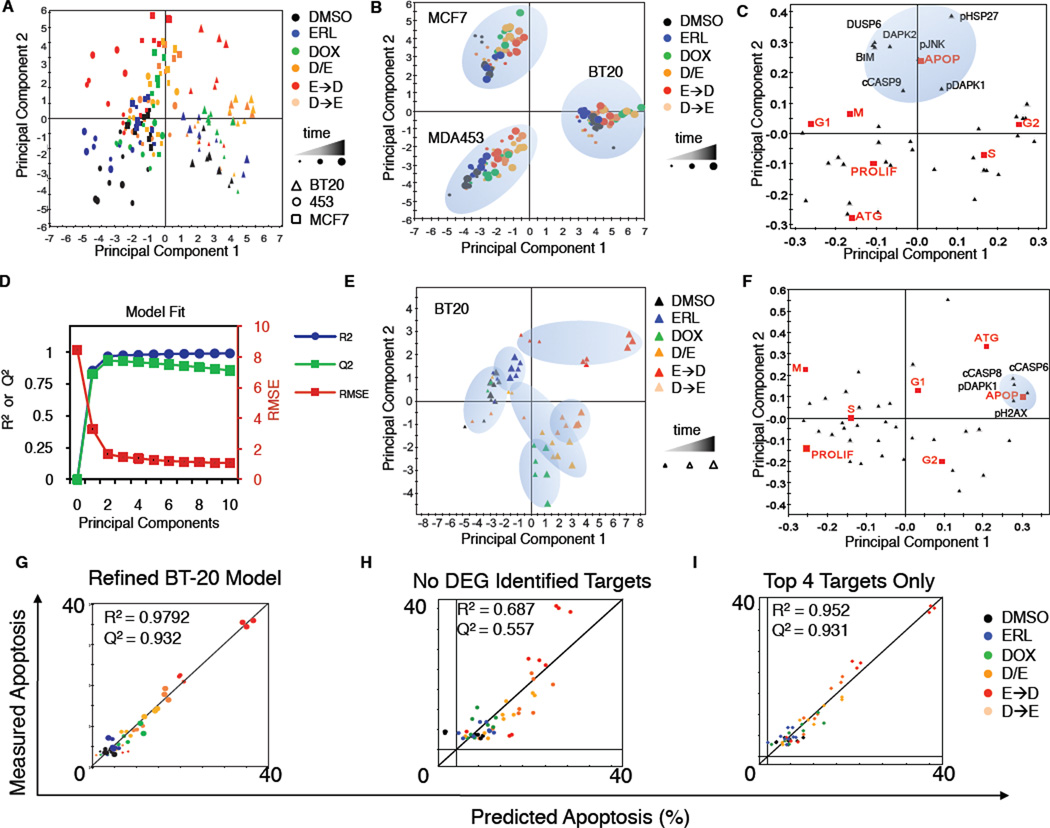
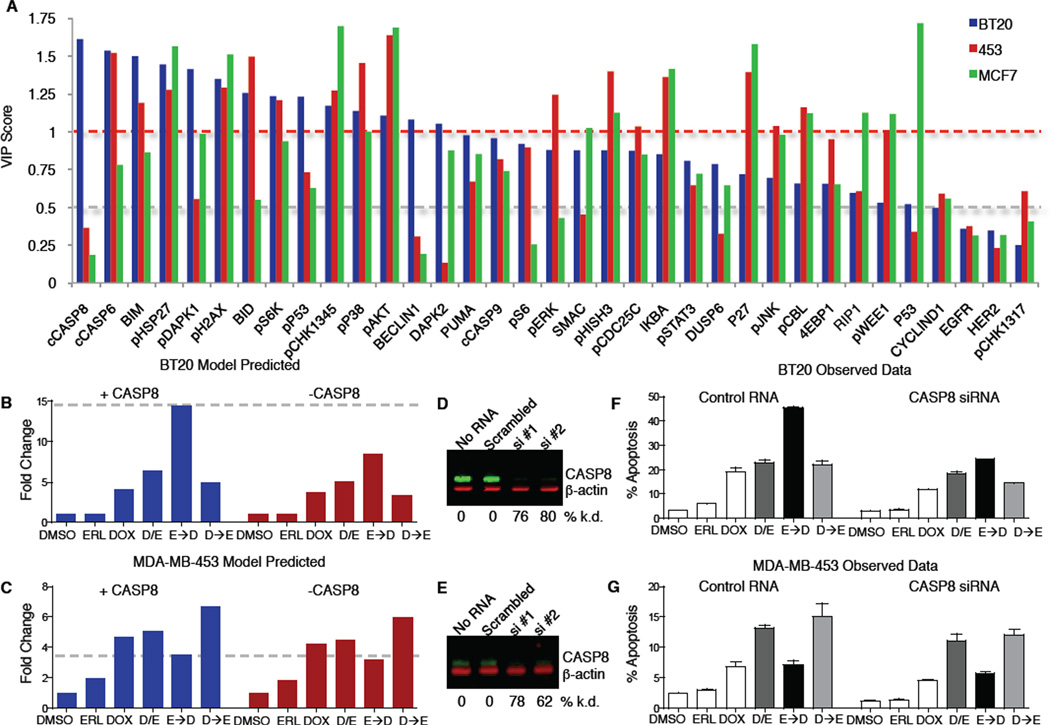
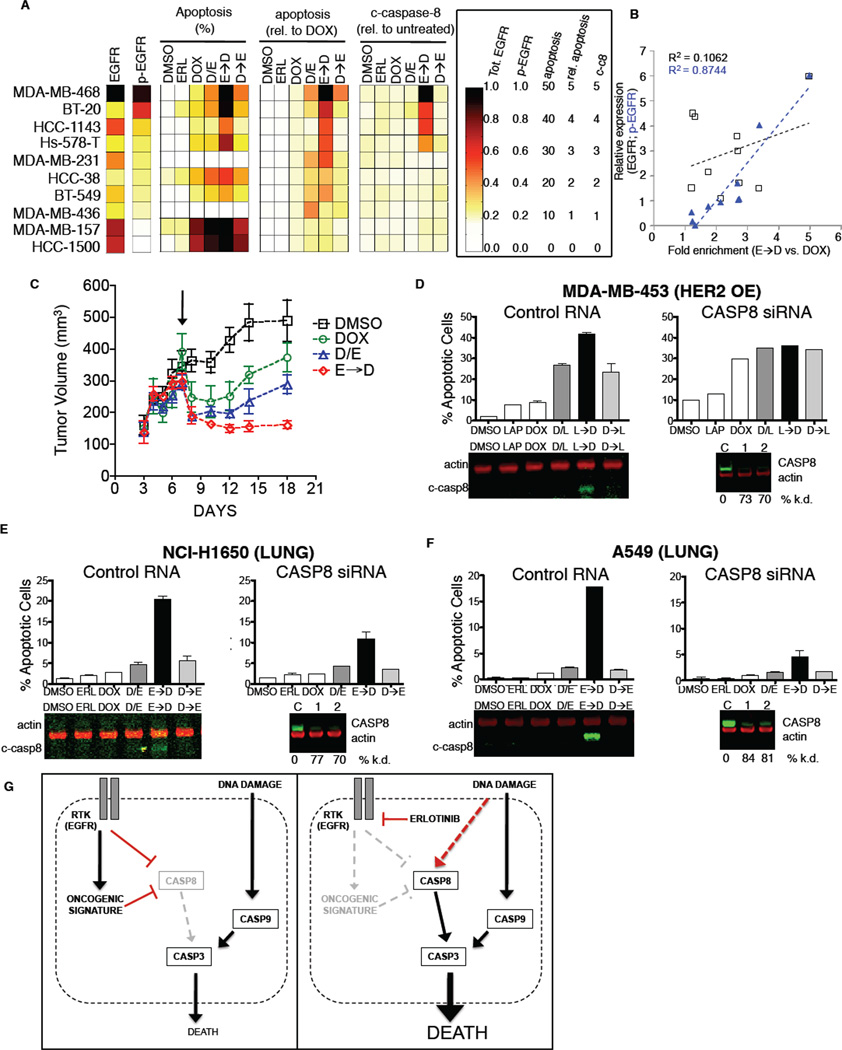
Comment in
-
Network medicine strikes a blow against breast cancer.Cell. 2012 May 11;149(4):731-3. doi: 10.1016/j.cell.2012.04.014. Cell. 2012. PMID: 22579276
-
Cell death. 1+1≠2.Nat Rev Cancer. 2012 Jun 14;12(7):449. doi: 10.1038/nrc3301. Nat Rev Cancer. 2012. PMID: 22695396
-
Throwing the kitchen sink at melanoma drug development.Pigment Cell Melanoma Res. 2012 Sep;25(5):543-4. doi: 10.1111/j.1755-148X.2012.01038.x. Pigment Cell Melanoma Res. 2012. PMID: 22812391 No abstract available.
-
LITERATURE Watch: implications for transplantation.Am J Transplant. 2013 Jan;13(1):3. doi: 10.1111/ajt.12072. Am J Transplant. 2013. PMID: 23279677 No abstract available.
Similar articles
-
Systems biology modeling reveals a possible mechanism of the tumor cell death upon oncogene inactivation in EGFR addicted cancers.PLoS One. 2011;6(12):e28930. doi: 10.1371/journal.pone.0028930. Epub 2011 Dec 14. PLoS One. 2011. PMID: 22194952 Free PMC article.
-
Alkylamino Phenol Derivative Induces Apoptosis by Inhibiting EGFR Signaling Pathway in Breast Cancer Cells.Anticancer Agents Med Chem. 2020;20(7):809-819. doi: 10.2174/1871520620666200213101407. Anticancer Agents Med Chem. 2020. PMID: 32053080
-
Dual inhibition of focal adhesion kinase and epidermal growth factor receptor pathways cooperatively induces death receptor-mediated apoptosis in human breast cancer cells.J Biol Chem. 2002 Oct 11;277(41):38978-87. doi: 10.1074/jbc.M205002200. Epub 2002 Aug 7. J Biol Chem. 2002. PMID: 12167618
-
Role of epidermal growth factor receptor in breast cancer.Breast Cancer Res Treat. 2012 Nov;136(2):331-45. doi: 10.1007/s10549-012-2289-9. Epub 2012 Oct 17. Breast Cancer Res Treat. 2012. PMID: 23073759 Free PMC article. Review.
-
Understanding EGFR Signaling in Breast Cancer and Breast Cancer Stem Cells: Overexpression and Therapeutic Implications.Asian Pac J Cancer Prev. 2016;17(2):445-53. doi: 10.7314/apjcp.2016.17.2.445. Asian Pac J Cancer Prev. 2016. PMID: 26925626 Review.
Cited by
-
MAPKAP Kinase-2 Drives Expression of Angiogenic Factors by Tumor-Associated Macrophages in a Model of Inflammation-Induced Colon Cancer.Front Immunol. 2021 Feb 23;11:607891. doi: 10.3389/fimmu.2020.607891. eCollection 2020. Front Immunol. 2021. PMID: 33708191 Free PMC article.
-
New insights into understanding the mechanisms, pathogenesis, and management of malignant mesotheliomas.Am J Pathol. 2013 Apr;182(4):1065-77. doi: 10.1016/j.ajpath.2012.12.028. Epub 2013 Feb 8. Am J Pathol. 2013. PMID: 23395095 Free PMC article. Review.
-
Anoikis evasion in inflammatory breast cancer cells is mediated by Bim-EL sequestration.Cell Death Differ. 2015 Aug;22(8):1275-86. doi: 10.1038/cdd.2014.209. Epub 2014 Dec 19. Cell Death Differ. 2015. PMID: 25526094 Free PMC article.
-
Amino Acid Conjugates of Aminothiazole and Aminopyridine as Potential Anticancer Agents: Synthesis, Molecular Docking and in vitro Evaluation.Drug Des Devel Ther. 2021 Apr 1;15:1459-1476. doi: 10.2147/DDDT.S297013. eCollection 2021. Drug Des Devel Ther. 2021. PMID: 33833504 Free PMC article.
-
Next-generation proteomics: towards an integrative view of proteome dynamics.Nat Rev Genet. 2013 Jan;14(1):35-48. doi: 10.1038/nrg3356. Epub 2012 Dec 4. Nat Rev Genet. 2013. PMID: 23207911 Review.
References
-
- Abeloff M, Wolff A, Weber B, Zaks T, Sacchini V, McCormick B. Cancer of the breast. In: Abeloff M, Armitage J, Niederhuber J, Kastan M, McKenna W, editors. Abeloff's Clinical Oncology. Maryland Heights, MO: Churchill Livingstone; 2008.
-
- Bosch A, Eroles P, Zaragoza R, Vina JR, Lluch A. Triple-negative breast cancer: molecular features, pathogenesis, treatment and current lines of research. Cancer Treat Rev. 2010;36:206–215. - PubMed
-
- Carey L, Rugo H, Marcom P, Irvin W, Ferraro M, Burrows E, He X, Perou C, Winer E. TBCRC 001: EGFR inhibition with cetuximab added to carboplatin in metastatic triple-negative (basal-like) breast cancer. J Clin Oncol. 2008;26
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
