

Autophagy is induced through the ROS-TP53-DRAM1 pathway in response to mitochondrial protein synthesis inhibition
- PMID: 22576012
- PMCID: PMC3429544
- DOI: 10.4161/auto.20250
Autophagy is induced through the ROS-TP53-DRAM1 pathway in response to mitochondrial protein synthesis inhibition
Abstract
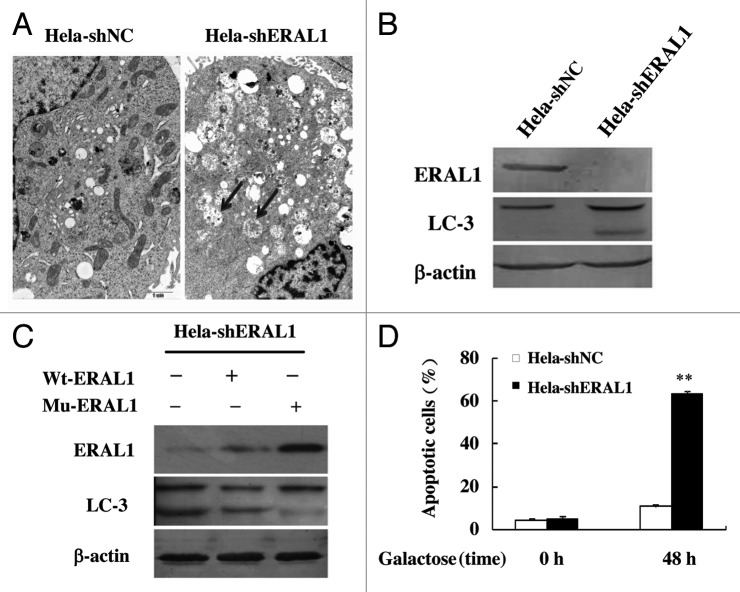
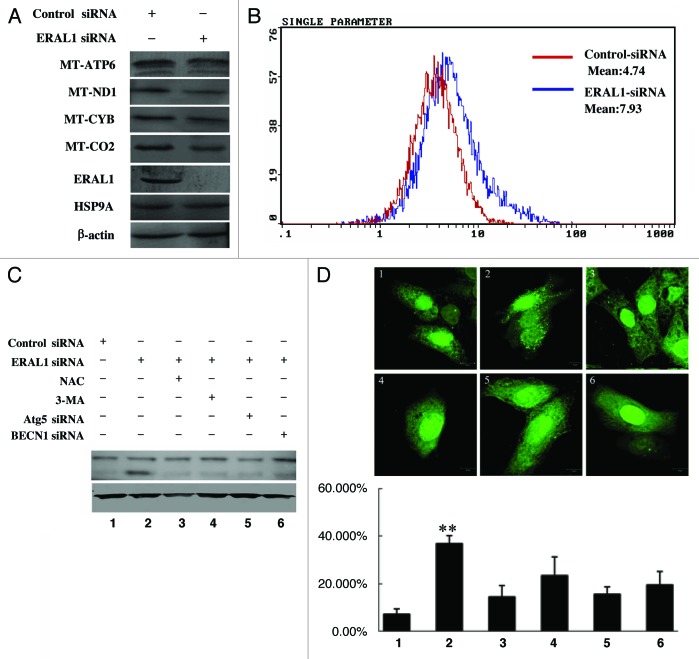
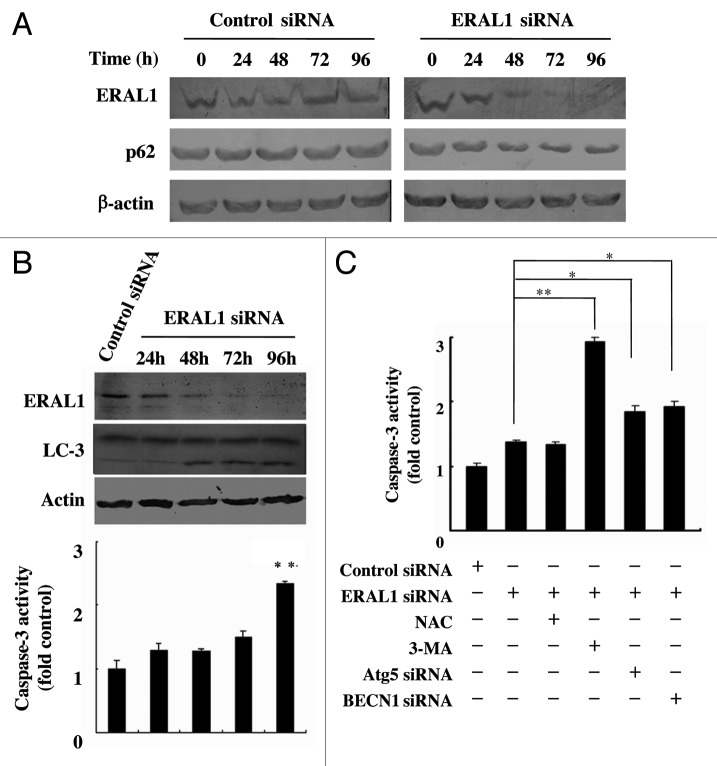
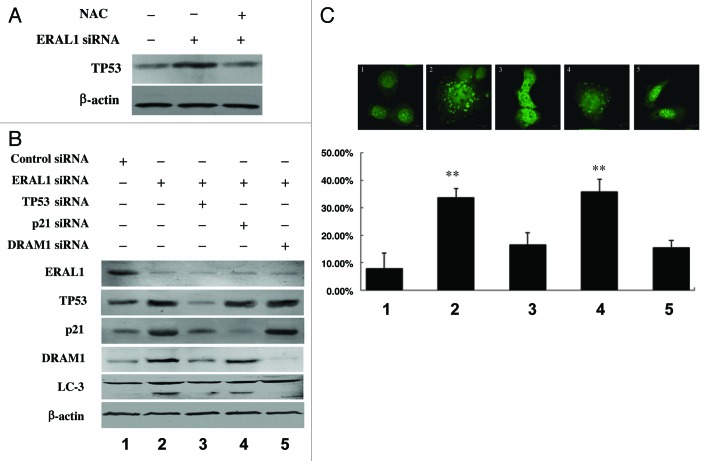
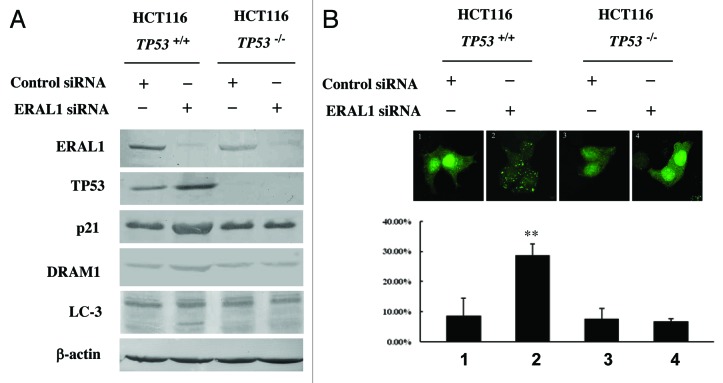
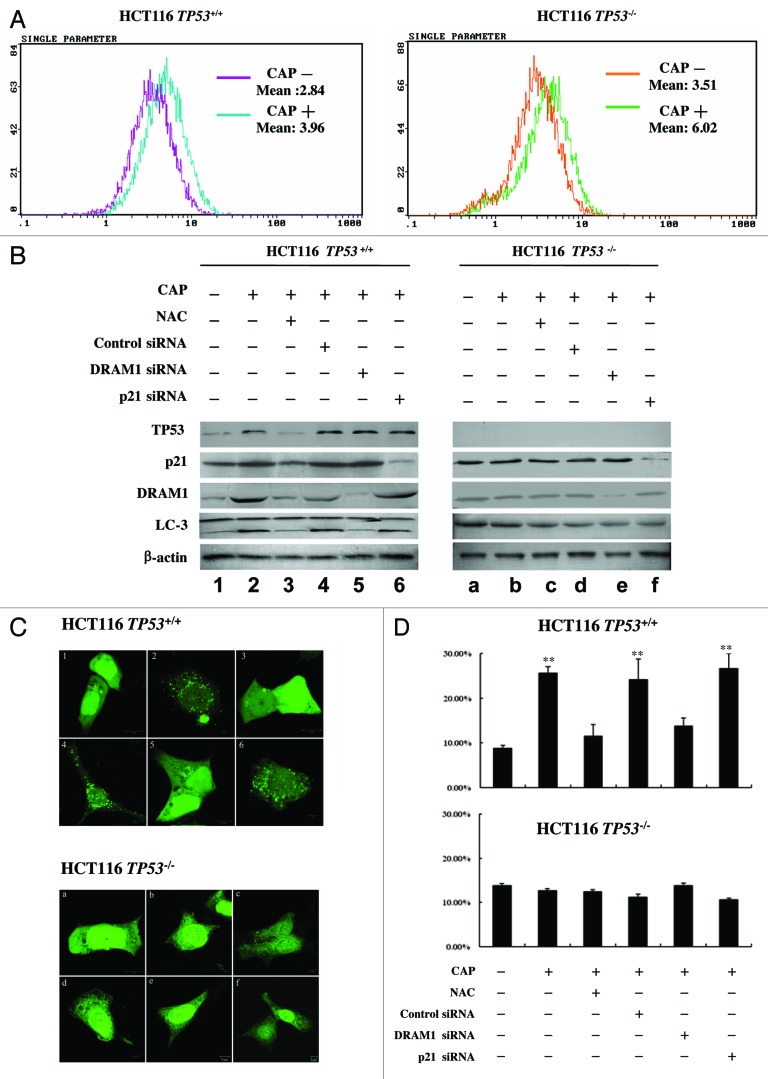
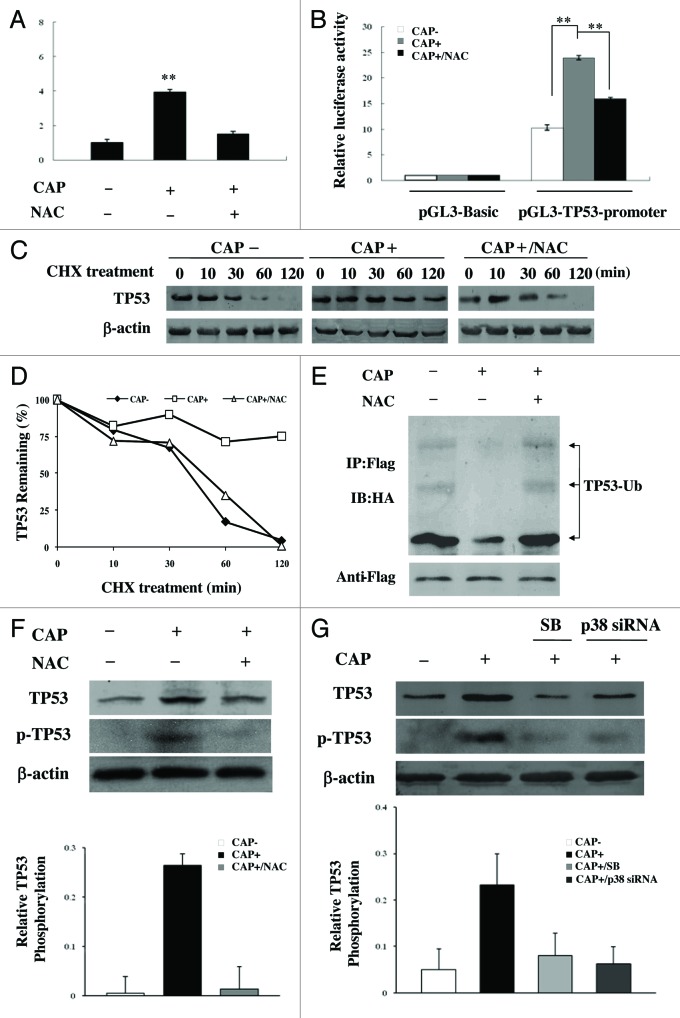
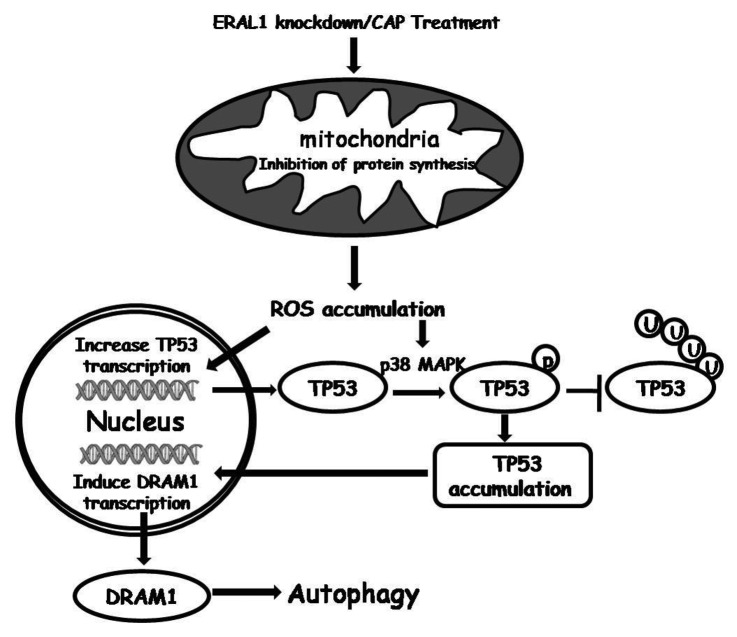
Mitoribosome in mammalian cells is responsible for synthesis of 13 mtDNA-encoded proteins, which are integral parts of four mitochondrial respiratory chain complexes (I, III, IV and V). ERAL1 is a nuclear-encoded GTPase important for the formation of the 28S small mitoribosomal subunit. Here, we demonstrate that knockdown of ERAL1 by RNA interference inhibits mitochondrial protein synthesis and promotes reactive oxygen species (ROS) generation, leading to autophagic vacuolization in HeLa cells. Cells that lack ERAL1 expression showed a significant conversion of LC3-I to LC3-II and an enhanced accumulation of autophagic vacuoles carrying the LC3 marker, all of which were blocked by the autophagy inhibitor 3-MA as well as by the ROS scavenger NAC. Inhibition of mitochondrial protein synthesis either by ERAL1 siRNA or chloramphenicol (CAP), a specific inhibitor of mitoribosomes, induced autophagy in HTC-116 TP53 (+/+) cells, but not in HTC-116 TP53 (-/-) cells, indicating that tumor protein 53 (TP53) is essential for the autophagy induction. The ROS elevation resulting from mitochondrial protein synthesis inhibition induced TP53 expression at transcriptional levels by enhancing TP53 promoter activity, and increased TP53 protein stability by suppressing TP53 ubiquitination through MAPK14/p38 MAPK-mediated TP53 phosphorylation. Upregulation of TP53 and its downstream target gene DRAM1, but not CDKN1A/p21, was required for the autophagy induction in ERAL1 siRNA or CAP-treated cells. Altogether, these data indicate that autophagy is induced through the ROS-TP53-DRAM1 pathway in response to mitochondrial protein synthesis inhibition.
Keywords: DRAM1; ERAL1; ROS; TP53; autophagy; chloramphenicol; mitoribosome.
Figures
Similar articles
-
DRAM1 regulates autophagy flux through lysosomes.PLoS One. 2013 May 17;8(5):e63245. doi: 10.1371/journal.pone.0063245. Print 2013. PLoS One. 2013. PMID: 23696801 Free PMC article.
-
Human ERAL1 is a mitochondrial RNA chaperone involved in the assembly of the 28S small mitochondrial ribosomal subunit.Biochem J. 2010 Sep 15;430(3):551-8. doi: 10.1042/BJ20100757. Biochem J. 2010. PMID: 20604745 Free PMC article.
-
Proteomic analysis revealed association of aberrant ROS signaling with suberoylanilide hydroxamic acid-induced autophagy in Jurkat T-leukemia cells.Autophagy. 2010 Aug;6(6):711-24. doi: 10.4161/auto.6.6.12397. Epub 2010 Aug 17. Autophagy. 2010. PMID: 20543569
-
The role of p53 in cell metabolism.Acta Pharmacol Sin. 2010 Sep;31(9):1208-12. doi: 10.1038/aps.2010.151. Epub 2010 Aug 23. Acta Pharmacol Sin. 2010. PMID: 20729871 Free PMC article. Review.
-
ROS and Autophagy: Interactions and Molecular Regulatory Mechanisms.Cell Mol Neurobiol. 2015 Jul;35(5):615-21. doi: 10.1007/s10571-015-0166-x. Epub 2015 Feb 27. Cell Mol Neurobiol. 2015. PMID: 25722131 Review.
Cited by
-
The p53 target DRAM1 modulates calcium homeostasis and ER stress by promoting contact between lysosomes and the ER through STIM1.Proc Natl Acad Sci U S A. 2024 Sep 24;121(39):e2400531121. doi: 10.1073/pnas.2400531121. Epub 2024 Sep 18. Proc Natl Acad Sci U S A. 2024. PMID: 39292746
-
DRAM1 plays a tumor suppressor role in NSCLC cells by promoting lysosomal degradation of EGFR.Cell Death Dis. 2020 Sep 17;11(9):768. doi: 10.1038/s41419-020-02979-9. Cell Death Dis. 2020. PMID: 32943616 Free PMC article.
-
Dysregulated autophagy in muscle precursor cells from humans with type 2 diabetes.Sci Rep. 2019 Jun 3;9(1):8169. doi: 10.1038/s41598-019-44535-2. Sci Rep. 2019. PMID: 31160616 Free PMC article.
-
DRAM1 protects neuroblastoma cells from oxygen-glucose deprivation/reperfusion-induced injury via autophagy.Int J Mol Sci. 2014 Oct 23;15(10):19253-64. doi: 10.3390/ijms151019253. Int J Mol Sci. 2014. PMID: 25342320 Free PMC article.
-
Role of CELF2 in ferroptosis: Potential targets for cancer therapy (Review).Int J Mol Med. 2023 Oct;52(4):88. doi: 10.3892/ijmm.2023.5291. Epub 2023 Aug 18. Int J Mol Med. 2023. PMID: 37594127 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous