

HIV-1 Vpr triggers mitochondrial destruction by impairing Mfn2-mediated ER-mitochondria interaction
- PMID: 22438978
- PMCID: PMC3306277
- DOI: 10.1371/journal.pone.0033657
HIV-1 Vpr triggers mitochondrial destruction by impairing Mfn2-mediated ER-mitochondria interaction
Abstract
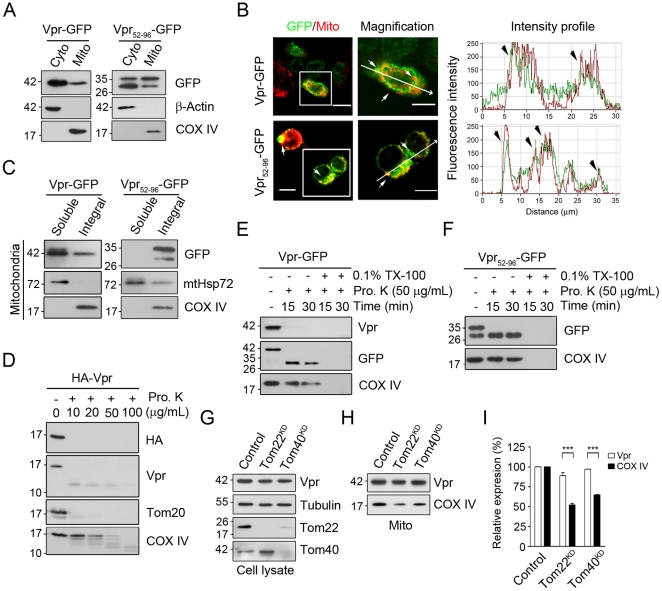
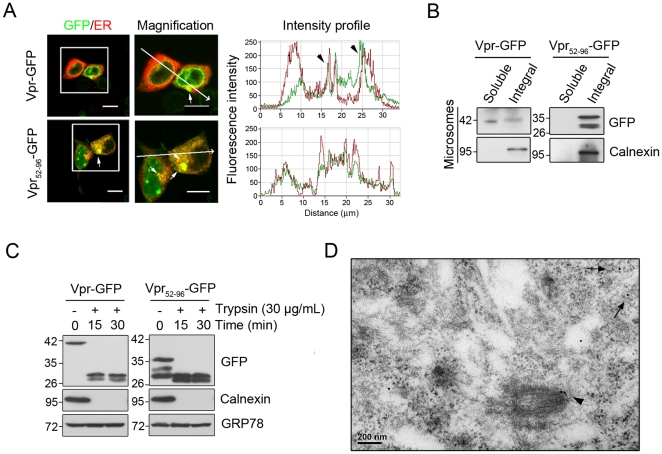
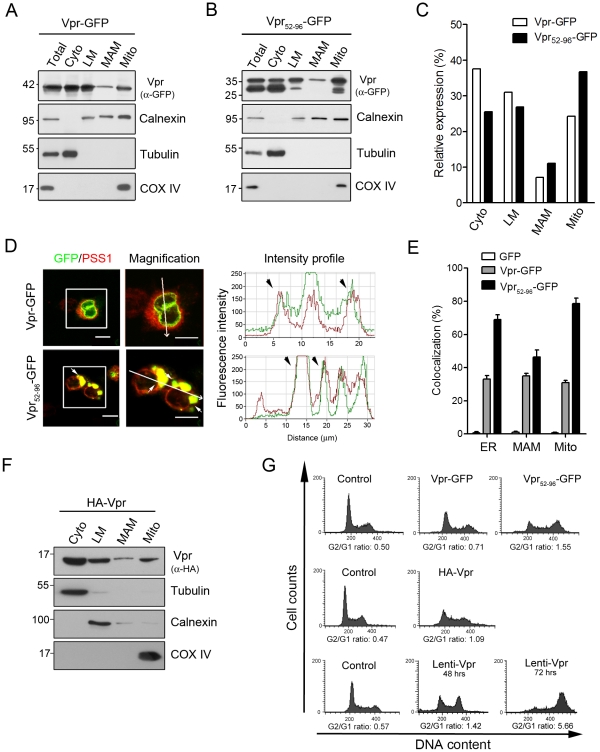
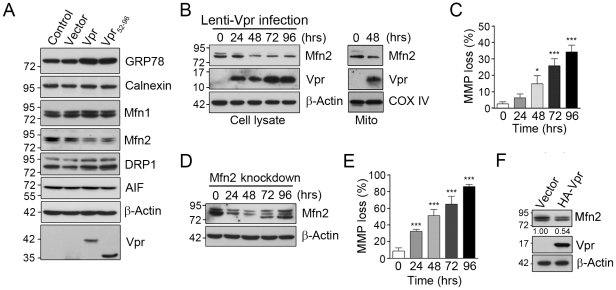
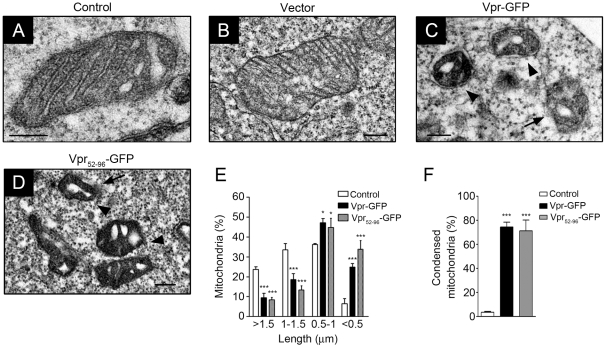
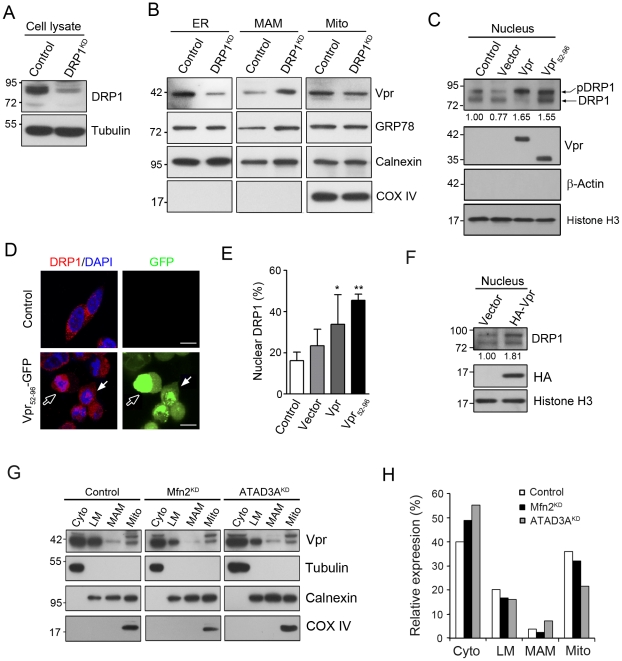
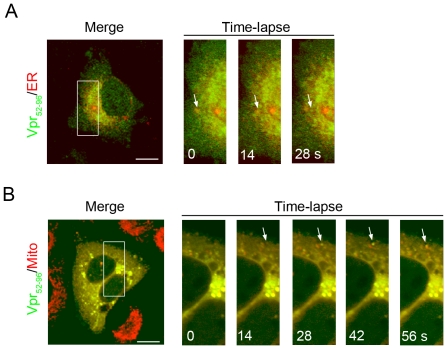
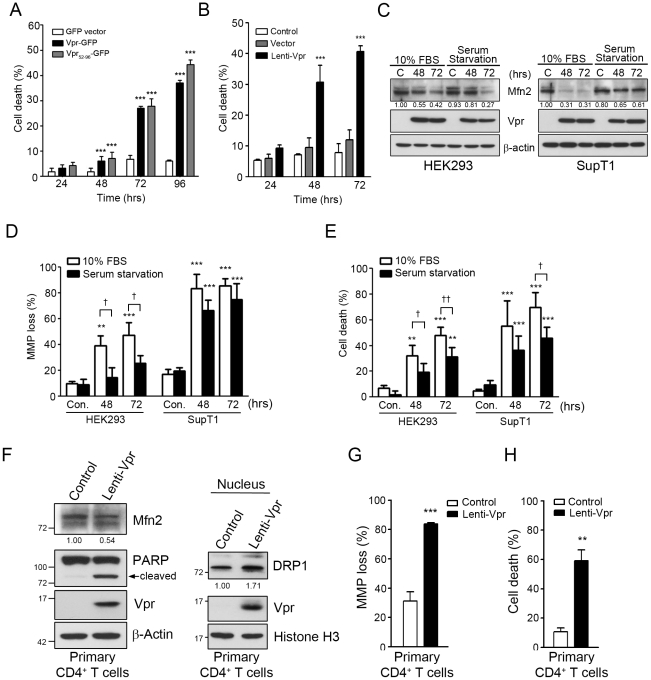
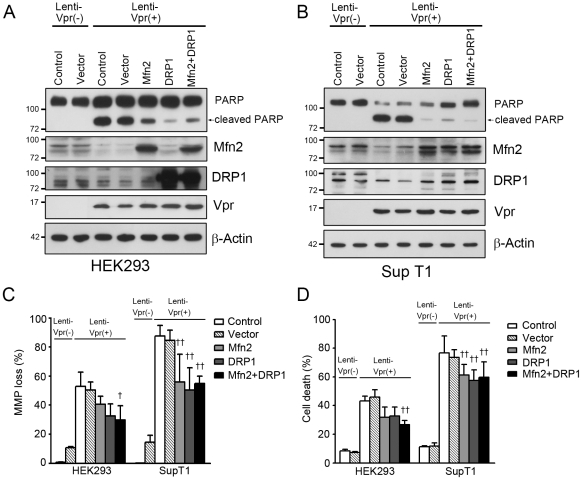
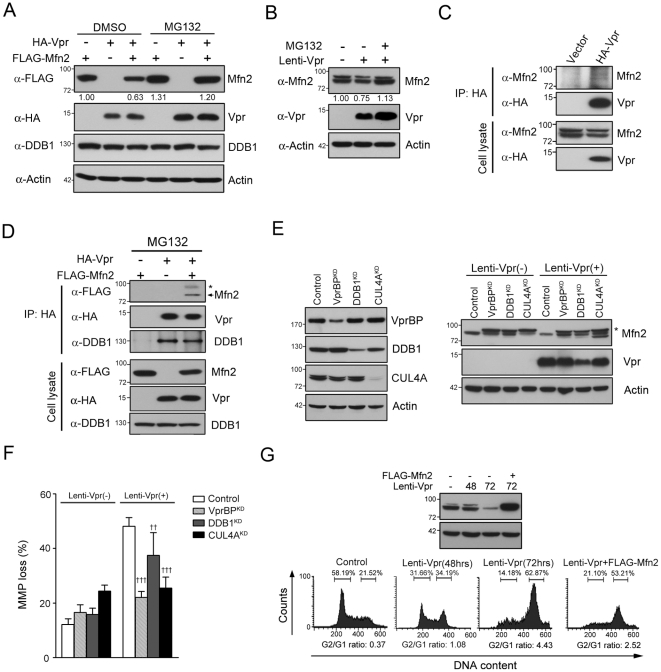
Human immunodeficiency virus 1 (HIV-1) viral protein R (Vpr) has been shown to induce host cell death by increasing the permeability of mitochondrial outer membrane (MOM). The mechanism underlying the damage to the mitochondria by Vpr, however, is not clearly illustrated. In this study, Vpr that is introduced, via transient transfection or lentivirus infection, into the human embryonic kidney cell line HEK293, human CD4(+) T lymphoblast cell line SupT1, or human primary CD4(+) T cells serves as the model system to study the molecular mechanism of Vpr-mediated HIV-1 pathogenesis. The results show that Vpr injures MOM and causes a loss in membrane potential (MMP) by posttranscriptionally reducing the expression of mitofusin 2 (Mfn2) via VprBP-DDB1-CUL4A ubiquitin ligase complex, gradually weakening MOM, and increasing mitochondrial deformation. Vpr also markedly decreases cytoplasmic levels of dynamin-related protein 1 (DRP1) and increases bulging in mitochondria-associated membranes (MAM), the specific regions of endoplasmic reticulum (ER) which form physical contacts with the mitochondria. Overexpression of Mfn2 and DRP1 significantly decreased the loss of MMP and apoptotic cell death caused by Vpr. Furthermore, by employing time-lapse confocal fluorescence microscopy, we identify the transport of Vpr protein from the ER, via MAM to the mitochondria. Taken together, our results suggest that Vpr-mediated cellular damage may occur on an alternative protein transport pathway from the ER, via MAM to the mitochondria, which are modulated by Mfn2 and DRP1.
Conflict of interest statement
Figures
Similar articles
-
Human MIEF1 recruits Drp1 to mitochondrial outer membranes and promotes mitochondrial fusion rather than fission.EMBO J. 2011 Jun 24;30(14):2762-78. doi: 10.1038/emboj.2011.198. EMBO J. 2011. PMID: 21701560 Free PMC article.
-
Receptor-mediated Drp1 oligomerization on endoplasmic reticulum.J Cell Biol. 2017 Dec 4;216(12):4123-4139. doi: 10.1083/jcb.201610057. Epub 2017 Nov 20. J Cell Biol. 2017. PMID: 29158231 Free PMC article.
-
Bax/Bak-dependent, Drp1-independent Targeting of X-linked Inhibitor of Apoptosis Protein (XIAP) into Inner Mitochondrial Compartments Counteracts Smac/DIABLO-dependent Effector Caspase Activation.J Biol Chem. 2015 Sep 4;290(36):22005-18. doi: 10.1074/jbc.M115.643064. Epub 2015 Jul 1. J Biol Chem. 2015. PMID: 26134559 Free PMC article.
-
New insights into the function and regulation of mitochondrial fission.Biochim Biophys Acta. 2013 May;1833(5):1256-68. doi: 10.1016/j.bbamcr.2013.02.002. Epub 2013 Feb 20. Biochim Biophys Acta. 2013. PMID: 23434681 Review.
-
Roles of mitochondrial ubiquitin ligase MITOL/MARCH5 in mitochondrial dynamics and diseases.J Biochem. 2014 May;155(5):273-9. doi: 10.1093/jb/mvu016. Epub 2014 Mar 9. J Biochem. 2014. PMID: 24616159 Review.
Cited by
-
Crosstalk between Dysfunctional Mitochondria and Proinflammatory Responses during Viral Infections.Int J Mol Sci. 2024 Aug 24;25(17):9206. doi: 10.3390/ijms25179206. Int J Mol Sci. 2024. PMID: 39273156 Free PMC article. Review.
-
Role of mitochondria in parvovirus pathology.PLoS One. 2014 Jan 21;9(1):e86124. doi: 10.1371/journal.pone.0086124. eCollection 2014. PLoS One. 2014. PMID: 24465910 Free PMC article.
-
From Entry to Egress: Strategic Exploitation of the Cellular Processes by HIV-1.Front Microbiol. 2020 Dec 4;11:559792. doi: 10.3389/fmicb.2020.559792. eCollection 2020. Front Microbiol. 2020. PMID: 33343516 Free PMC article. Review.
-
Positive-sense RNA viruses reveal the complexity and dynamics of the cellular and viral epitranscriptomes during infection.Nucleic Acids Res. 2018 Jun 20;46(11):5776-5791. doi: 10.1093/nar/gky029. Nucleic Acids Res. 2018. PMID: 29373715 Free PMC article.
-
Mitochondrial Functions in Infection and Immunity.Trends Cell Biol. 2020 Apr;30(4):263-275. doi: 10.1016/j.tcb.2020.01.006. Epub 2020 Feb 11. Trends Cell Biol. 2020. PMID: 32200805 Free PMC article. Review.
References
-
- Emerman M, Malim MH. HIV-1 regulatory/accessory genes: keys to unraveling viral and host cell biology. Science. 1998;280:1880–1884. - PubMed
-
- Bukrinsky M, Adzhubei A. Viral protein R of HIV-1. Rev Med Virol. 1999;9:39–49. - PubMed
-
- Muthumani K, Choo AY, Hwang DS, Chattergoon MA, Dayes NN, et al. Mechanism of HIV-1 viral protein R-induced apoptosis. Biochem Biophys Res Commun. 2003;304:583–592. - PubMed
-
- Andersen JL, Planelles V. The role of Vpr in HIV-1 pathogenesis. Curr HIV Res. 2005;3:43–51. - PubMed
-
- Muthumani K, Choo AY, Premkumar A, Hwang DS, Thieu KP, et al. Human immunodeficiency virus type 1 (HIV-1) Vpr-regulated cell death: insights into mechanism. Cell Death Differ. 2005;12(Suppl 1):962–970. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
