

Evolutionary dynamics of Staphylococcus aureus during progression from carriage to disease
- PMID: 22393007
- PMCID: PMC3311376
- DOI: 10.1073/pnas.1113219109
Evolutionary dynamics of Staphylococcus aureus during progression from carriage to disease
Abstract
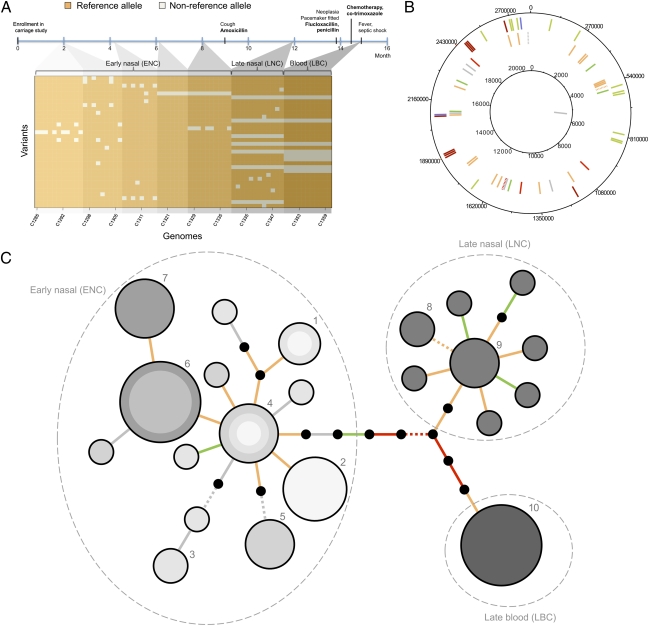
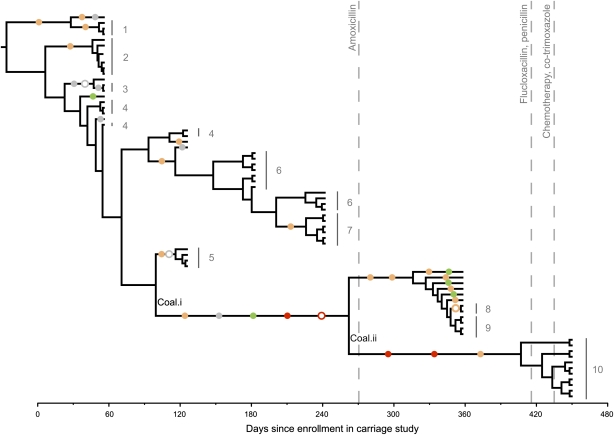
Whole-genome sequencing offers new insights into the evolution of bacterial pathogens and the etiology of bacterial disease. Staphylococcus aureus is a major cause of bacteria-associated mortality and invasive disease and is carried asymptomatically by 27% of adults. Eighty percent of bacteremias match the carried strain. However, the role of evolutionary change in the pathogen during the progression from carriage to disease is incompletely understood. Here we use high-throughput genome sequencing to discover the genetic changes that accompany the transition from nasal carriage to fatal bloodstream infection in an individual colonized with methicillin-sensitive S. aureus. We found a single, cohesive population exhibiting a repertoire of 30 single-nucleotide polymorphisms and four insertion/deletion variants. Mutations accumulated at a steady rate over a 13-mo period, except for a cluster of mutations preceding the transition to disease. Although bloodstream bacteria differed by just eight mutations from the original nasally carried bacteria, half of those mutations caused truncation of proteins, including a premature stop codon in an AraC-family transcriptional regulator that has been implicated in pathogenicity. Comparison with evolution in two asymptomatic carriers supported the conclusion that clusters of protein-truncating mutations are highly unusual. Our results demonstrate that bacterial diversity in vivo is limited but nonetheless detectable by whole-genome sequencing, enabling the study of evolutionary dynamics within the host. Regulatory or structural changes that occur during carriage may be functionally important for pathogenesis; therefore identifying those changes is a crucial step in understanding the biological causes of invasive bacterial disease.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Within-host evolution of Staphylococcus aureus during asymptomatic carriage.PLoS One. 2013 May 1;8(5):e61319. doi: 10.1371/journal.pone.0061319. Print 2013. PLoS One. 2013. PMID: 23658690 Free PMC article.
-
Evolution of the Staphylococcus argenteus ST2250 Clone in Northeastern Thailand Is Linked with the Acquisition of Livestock-Associated Staphylococcal Genes.mBio. 2017 Jul 5;8(4):e00802-17. doi: 10.1128/mBio.00802-17. mBio. 2017. PMID: 28679748 Free PMC article.
-
Natural population dynamics and expansion of pathogenic clones of Staphylococcus aureus.J Clin Invest. 2004 Dec;114(12):1732-40. doi: 10.1172/JCI23083. J Clin Invest. 2004. PMID: 15599398 Free PMC article.
-
The evolution of vancomycin intermediate Staphylococcus aureus (VISA) and heterogenous-VISA.Infect Genet Evol. 2014 Jan;21:575-82. doi: 10.1016/j.meegid.2013.03.047. Epub 2013 Apr 6. Infect Genet Evol. 2014. PMID: 23567819 Review.
-
Population genetics and the evolution of virulence in Staphylococcus aureus.Infect Genet Evol. 2014 Jan;21:554-62. doi: 10.1016/j.meegid.2013.04.026. Epub 2013 Apr 27. Infect Genet Evol. 2014. PMID: 23628638 Review.
Cited by
-
Synergistic Antibiofilm Activity between Synthetic Peptides and Ciprofloxacin against Staphylococcus aureus.Pathogens. 2022 Aug 31;11(9):995. doi: 10.3390/pathogens11090995. Pathogens. 2022. PMID: 36145427 Free PMC article.
-
Whole-genome sequencing of uropathogenic Escherichia coli reveals long evolutionary history of diversity and virulence.Infect Genet Evol. 2015 Aug;34:244-50. doi: 10.1016/j.meegid.2015.06.023. Epub 2015 Jun 23. Infect Genet Evol. 2015. PMID: 26112070 Free PMC article.
-
Improved understanding of factors driving methicillin-resistant Staphylococcus aureus epidemic waves.Clin Epidemiol. 2013 Jul 4;5:205-17. doi: 10.2147/CLEP.S37071. Print 2013. Clin Epidemiol. 2013. PMID: 23861600 Free PMC article.
-
Pneumococcal genetic variability in age-dependent bacterial carriage.Elife. 2022 Jul 26;11:e69244. doi: 10.7554/eLife.69244. Elife. 2022. PMID: 35881438 Free PMC article.
-
Rapid whole-genome sequencing for detection and characterization of microorganisms directly from clinical samples.J Clin Microbiol. 2014 Jan;52(1):139-46. doi: 10.1128/JCM.02452-13. Epub 2013 Oct 30. J Clin Microbiol. 2014. PMID: 24172157 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
