

The nuclear cofactor RAC3/AIB1/SRC-3 enhances Nrf2 signaling by interacting with transactivation domains
- PMID: 22370642
- PMCID: PMC3538952
- DOI: 10.1038/onc.2012.59
The nuclear cofactor RAC3/AIB1/SRC-3 enhances Nrf2 signaling by interacting with transactivation domains
Abstract
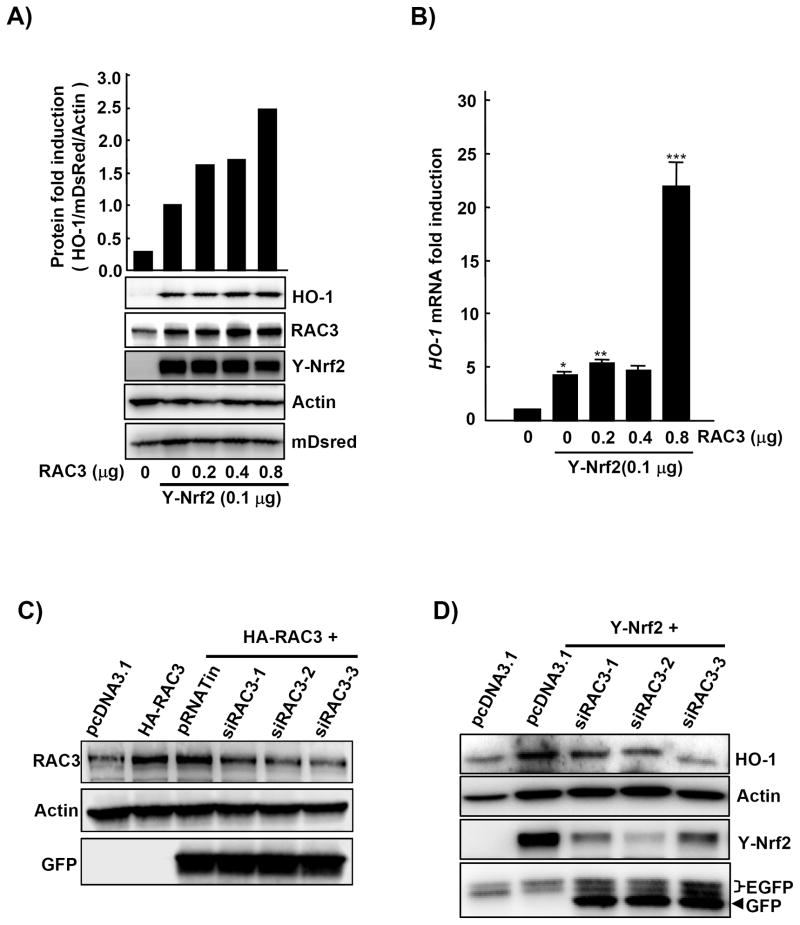
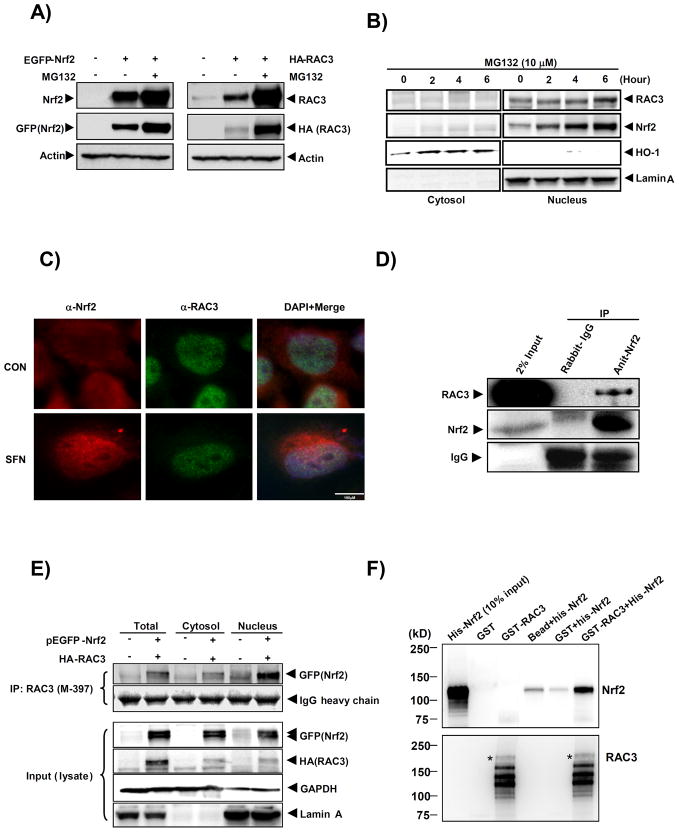
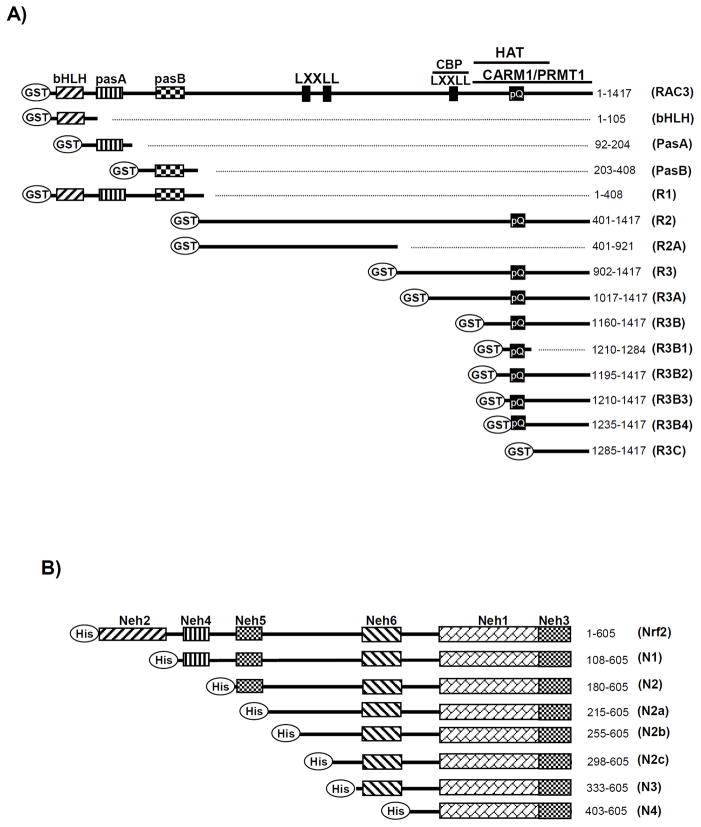
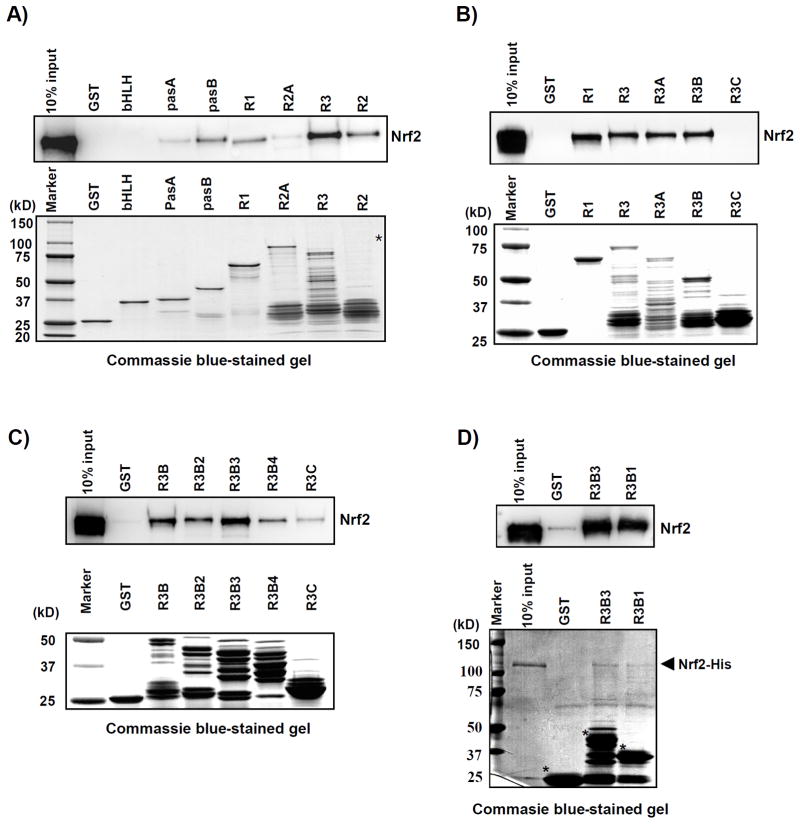
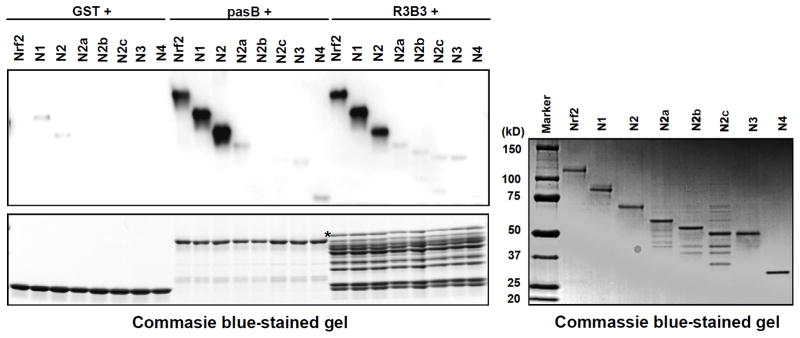
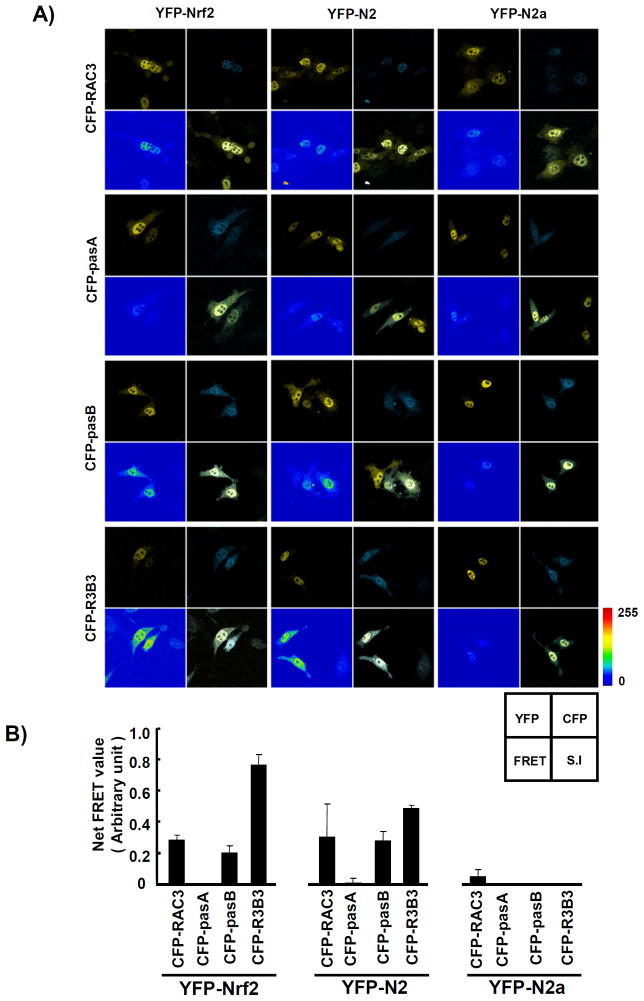
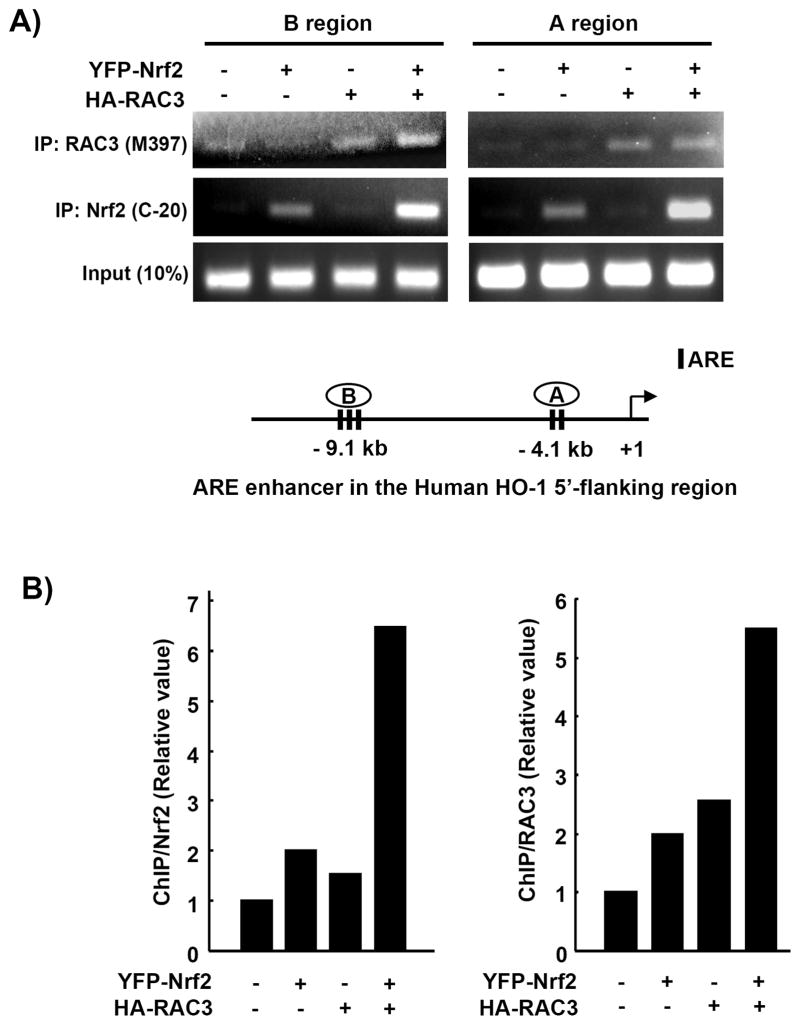
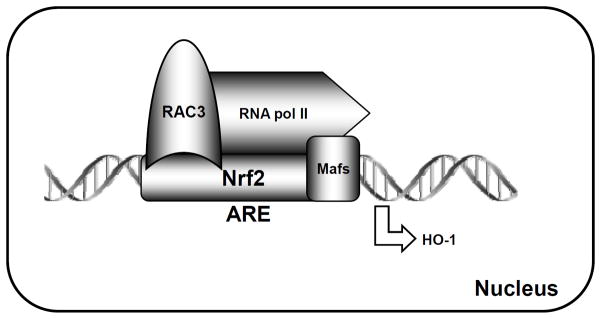
Nuclear factor erythroid 2-related factor 2 (Nrf2, NM 006164, 605 AA) is essential for the antioxidant responsive element (ARE)-mediated expression of a group of detoxifying antioxidant genes that detoxify carcinogens and protect against oxidative stress. Several proteins have been identified as Nrf2-interacting molecules. In this study, we found that the overexpression of receptor-associated coactivator 3 (RAC3)/AIB-1/steroid receptor coactivator-3, a nuclear coregulator and oncogene frequently amplified in human breast cancers, induced heme oxygenase-1 (HO-1) through Nrf2 transactivation in HeLa cells. Next, we determined the interaction between RAC3 and Nrf2 proteins using a co-immunoprecipitation assay and fluorescence resonance energy transfer analysis. The results showed that RAC3 bound directly to the Nrf2 protein in the nucleus. Subsequently, we identified the interacting domains of Nrf2 and RAC3 using a glutathione S-transferase pull-down assay. The results showed that both the N-terminal RAC3-pasB and C-terminal RAC3-R3B3 domains were tightly bound to the Neh4 and Neh5 transactivation domains. Furthermore, chromatin immunoprecipitation showed that RAC3 bound tightly to the ARE enhancer region of the HO-1 promoter via Nrf2 binding. These data suggest that Nrf2 activation is modulated and directly controlled through interactions with the RAC3 protein in HeLa cells.
Figures
Similar articles
-
Regulation of Nrf2 transactivation domain activity by p160 RAC3/SRC3 and other nuclear co-regulators.J Biochem Mol Biol. 2006 May 31;39(3):304-10. doi: 10.5483/bmbrep.2006.39.3.304. J Biochem Mol Biol. 2006. PMID: 16756760
-
Identification and functional studies of a new Nrf2 partner IQGAP1: a critical role in the stability and transactivation of Nrf2.Antioxid Redox Signal. 2013 Jul 10;19(2):89-101. doi: 10.1089/ars.2012.4586. Epub 2012 Sep 17. Antioxid Redox Signal. 2013. PMID: 22793650 Free PMC article.
-
Nrf2 Neh5 domain is differentially utilized in the transactivation of cytoprotective genes.Biochem J. 2007 Jun 15;404(3):459-66. doi: 10.1042/BJ20061611. Biochem J. 2007. PMID: 17313370 Free PMC article.
-
SRC-3/AIB1: transcriptional coactivator in oncogenesis.Acta Pharmacol Sin. 2006 Apr;27(4):387-94. doi: 10.1111/j.1745-7254.2006.00315.x. Acta Pharmacol Sin. 2006. PMID: 16539836 Review.
-
Synergistic Interaction Between Heme Oxygenase (HO) and Nuclear-Factor E2- Related Factor-2 (Nrf2) against Oxidative Stress in Cardiovascular Related Diseases.Curr Pharm Des. 2017;23(10):1465-1470. doi: 10.2174/1381612823666170113153818. Curr Pharm Des. 2017. PMID: 28088909 Review.
Cited by
-
Camellia japonica Root Extract Increases Antioxidant Genes by Induction of NRF2 in HeLa Cells.Plants (Basel). 2022 Oct 29;11(21):2914. doi: 10.3390/plants11212914. Plants (Basel). 2022. PMID: 36365366 Free PMC article.
-
The NRF2/KEAP1 Axis in the Regulation of Tumor Metabolism: Mechanisms and Therapeutic Perspectives.Biomolecules. 2020 May 20;10(5):791. doi: 10.3390/biom10050791. Biomolecules. 2020. PMID: 32443774 Free PMC article. Review.
-
Dysregulation of Nrf2 in Hepatocellular Carcinoma: Role in Cancer Progression and Chemoresistance.Cancers (Basel). 2018 Dec 3;10(12):481. doi: 10.3390/cancers10120481. Cancers (Basel). 2018. PMID: 30513925 Free PMC article. Review.
-
Nuclear TIGAR mediates an epigenetic and metabolic autoregulatory loop via NRF2 in cancer therapeutic resistance.Acta Pharm Sin B. 2022 Apr;12(4):1871-1884. doi: 10.1016/j.apsb.2021.10.015. Epub 2021 Oct 21. Acta Pharm Sin B. 2022. PMID: 35847493 Free PMC article.
-
Coordinated regulation of Nrf2 and histone H3 serine 10 phosphorylation in arsenite-activated transcription of the human heme oxygenase-1 gene.Biochim Biophys Acta. 2015 Oct;1849(10):1277-88. doi: 10.1016/j.bbagrm.2015.08.004. Epub 2015 Aug 18. Biochim Biophys Acta. 2015. PMID: 26291278 Free PMC article.
References
-
- Alam J, Stewart D, Touchard C, Boinapally S, Choi AM, Cook JL. J Biol Chem. 1999;274:26071–8. - PubMed
-
- Anzick SL, Kononen J, Walker RL, Azorsa DO, Tanner MM, Guan XY, Sauter G, Kallioniemi OP, Trent JM, Meltzer PS. Science. 1997;277:965–8. - PubMed
-
- Arimura A, vn Peer M, Schroder AJ, Rothman PB. J Biol Chem. 2004;279:31105–12. - PubMed
-
- Blank V. J Mol Biol. 2008;376:913–25. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
